

{kind=link}
An FDA-approved diabetes drug shows early signs of promise against the most common genetic form of amyotrophic lateral sclerosis, a devastating neurological condition that causes paralysis.
ALS is a progressive disease that affects neurons in the brain and spinal cord. Motor neurons transmit signals from our brain to our muscles and allow us to move. ALS causes these motor neurons to die, resulting in the loss of a patient’s ability to speak, eat, move and breathe. Notable ALS patients include New York Yankees baseball star Lou Gehrig (the disease is often called Lou Gehrig’s disease), physicist Stephen Hawking and New Orleans Saints football star Steve Gleason. There are currently more than 30,000 cases of ALS in the United States, and life expectancy after diagnosis is typically 2 to 5 years. There is currently no cure for ALS.
I am a scientist who studies neurological diseases that run in families, and I have been working hard to find a treatment to stop ALS. Our team has made a discovery, detailed in a scientific study and highlighted in a commentary by Nobel Prize winner Michael Rosbash, that paves the way for further research for improving disease in a genetic type of ALS caused by a mutation in a gene with the unwieldy name chromosome 9 open reading frame 72 (C9orf72), based on its location on chromosome 9. In addition to ALS, mutations in this gene can also cause frontotemporal dementia, which can cause apathy, loss of emotional control and cognitive decline. Some patients with the C9orf72 mutation develop ALS, others develop frontotemporal dementia and some develop both. Together, these diseases are referred to here as C9-ALS/FTD.
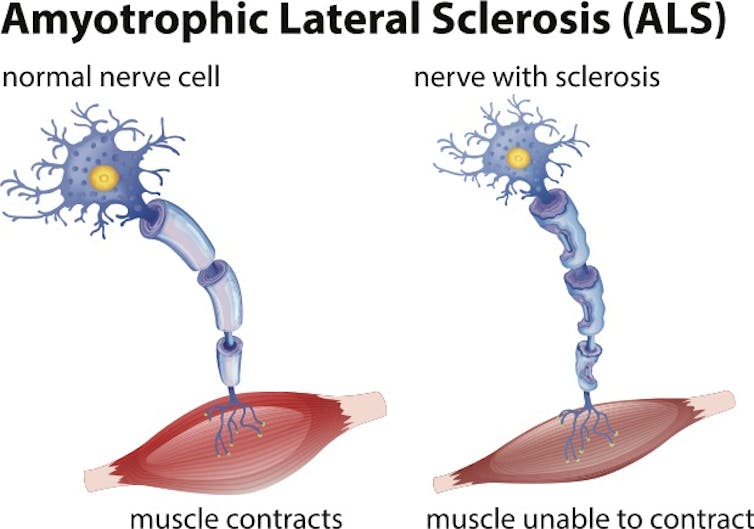
Repeating theme in ALS and other neurodegenerative diseases
I have been focusing on C9-ALS, which is the most common genetic type of ALS which is caused by a mutation in the C9orf72 gene. The mutation occurs when six letters of DNA that make up part of the gene’s genetic code – GGGGCC – are repeated hundreds of extra times. It is as if a single word is repeated hundreds of times in the same sentence.
[Deep knowledge, daily. Sign up for The Conversation’s newsletter.]
The gene mutation that causes C9-ALS is part of a much larger family of diseases that are caused by similar expansions of short repeated segments of DNA.
Repeat expansion mutations were first discovered in 1991 as the cause of Fragile X syndrome and spinal bulbar muscular atrophy, and this type of mutation is now known to cause more than 50 different neurologic diseases.
Over the past 30 years, I have been studying these types of expansion mutations, including those that cause a disease called spinocerebellar ataxia type 8 that affects coordination and the muscle disease myotonic dystrophy type 2. I have been particularly interested in understanding how these gene mutations work, and there have been some big surprises.
One mutation, two RNAs and six unexpected proteins
Typically, a gene encoded in DNA makes a single copy of an intermediate molecule called RNA, which the cell uses to manufacture a protein. Also, portions of genes are typically not expressed as proteins. The repeat expansion mutation in C9-ALS and many other neurological diseases occurs in these gene regions not expected to produce proteins. But, when a repeat expansion is present, the mutated region produces up to six unexpected and toxic proteins.
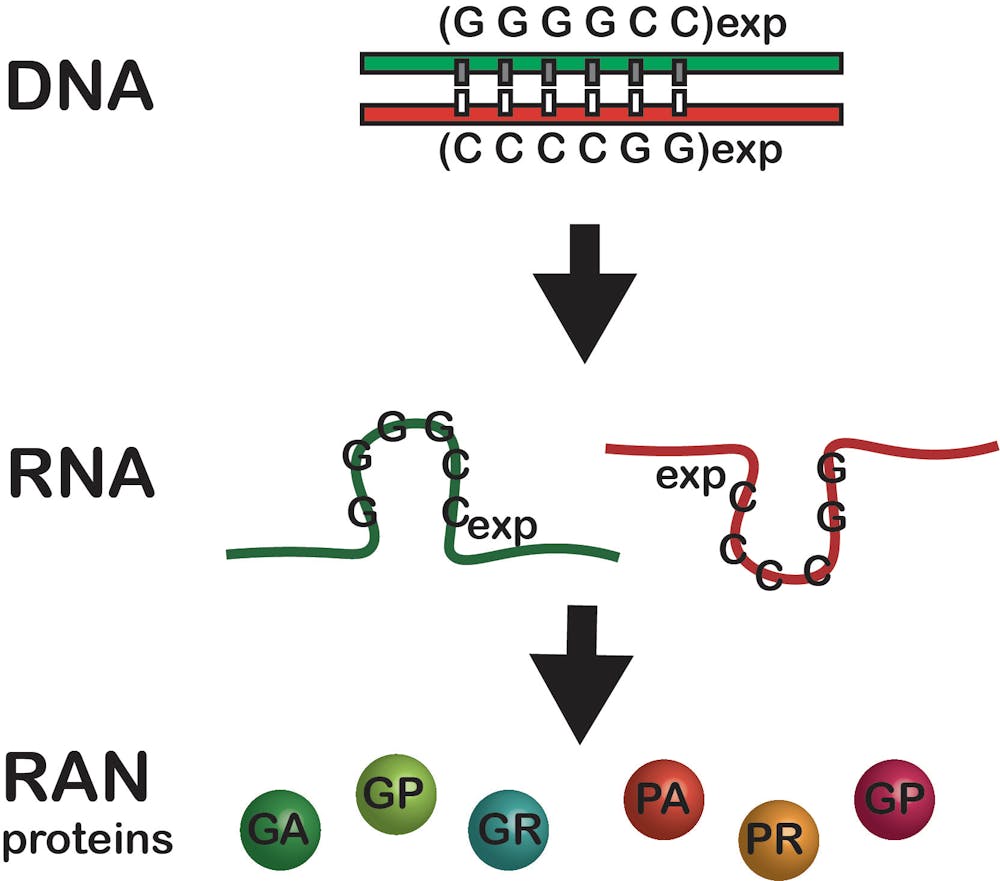
In 2006, we started to unravel how this happens by discovering that the spinocerebellar ataxia type 8 repeat expansion mutation produces two RNAs instead of just one. This double-the-trouble scenario was also found in myotonic dystrophy type 1 and is now known to occur for many repeat expansion disorders.
In 2011, we discovered that these repeat-expansion mutations also break the previously established dogma that a start signal embedded in the genetic code is required to make proteins. In contrast, we showed that when a repeat expansion mutation is present, proteins are produced without a start signal and extra, unexpected proteins are made – up to six for each expansion mutation.
We called these rogue proteins repeat associated non-AUG (RAN) proteins. These proteins accumulate in neurons and other brain cells, damaging them and causing disease. RAN proteins have now been found in 10 different repeat expansion diseases, including Huntington’s disease and Fragile X tremor ataxia syndrome.
In C9-ALS mice, we have shown that destroying RAN proteins using antibodies in mice improves lifespan, the survival of motor neurons and other key aspects of the disease.
In the newly published study in the Proceedings of the National Academy of Sciences, our team discovered a cellular switch that, when turned on, hijacks the cell and forces it into making RAN proteins. RNA copies of the expansion mutations turn on this switch, called the protein kinase R pathway.
Turning off the protein kinase R pathway switch blocks RAN protein production for multiple types of disease-causing repeat expansions, making protein kinase R a possible Achilles heel for RAN protein diseases. These results had me and my colleague Tao Zu, a research assistant professor, tremendously excited.
Metformin shows promise for C9-ALS/FTD and other expansion diseases
I decided that we should test the FDA-approved diabetes drug metformin after hearing Nahum Sonenberg, a longtime collaborator, present data showing that this drug improved disease in mice with Fragile X syndrome, a disease involving a missing protein. Even though Fragile X disrupts protein production in a completely different way, I thought that because metformin normalized protein production in Fragile X syndrome, maybe it could do the same for RAN protein diseases.
Although it was a long shot, I asked Dr. Zu to test metformin in cells to see if the drug would lower RAN protein levels. The results in cells very clearly showed that it did. We went on to show that metformin inhibits protein kinase R, reduces RAN proteins and improves disease in C9-ALS/FTD mice. It is important to emphasize that this approach is thought to work for this particular genetic form of ALS and frontotemporal dementia because the C9orf72 mutation makes RAN proteins. In a previous research study conducted by a different group, metformin treatment was not effective in mice with a different form of ALS that does not produce RAN proteins.
Typically, it takes a decade or more to move promising research from the lab to the clinic. Metformin was introduced in 1957 in France and approved in 1995 in the United States. Because metformin is widely used as a safe and effective treatment for Type 2 diabetes with few side effects, we can skip the arduous drug-development process and immediately test if the benefits of metformin treatment in mice are also found in people with C9-ALS.
My colleagues and I at the University of Florida have already started a Phase 2 open-label trial to test the effects of metformin in C9-ALS patients. In this first open-label trial, in which everyone will receive the drug, we will be testing to see if the drug is safe for patients with C9-ALS and if it lowers RAN protein levels in the cerebrospinal fluid.
If the results are promising, the next step would be to test the potential benefits of the drug in a larger, placebo-controlled trial. We are especially excited about metformin as a potential treatment for C9-ALS/FTD and other repeat expansion disorders, because by reducing RAN proteins, it could address a fundamental problem common to many of these diseases.