
Imagine the thrill of discovery when more than 10 years of research on the origin of a common genetic disease, cystic fibrosis (CF), results in tracing it to a group of distinct but mysterious Europeans who lived about 5,000 years ago.
CF is the most common, potentially lethal, inherited disease among Caucasians – about one in 40 carry the so-called F508del mutation. Typically only beneficial mutations, which provide a survival advantage, spread widely through a population.
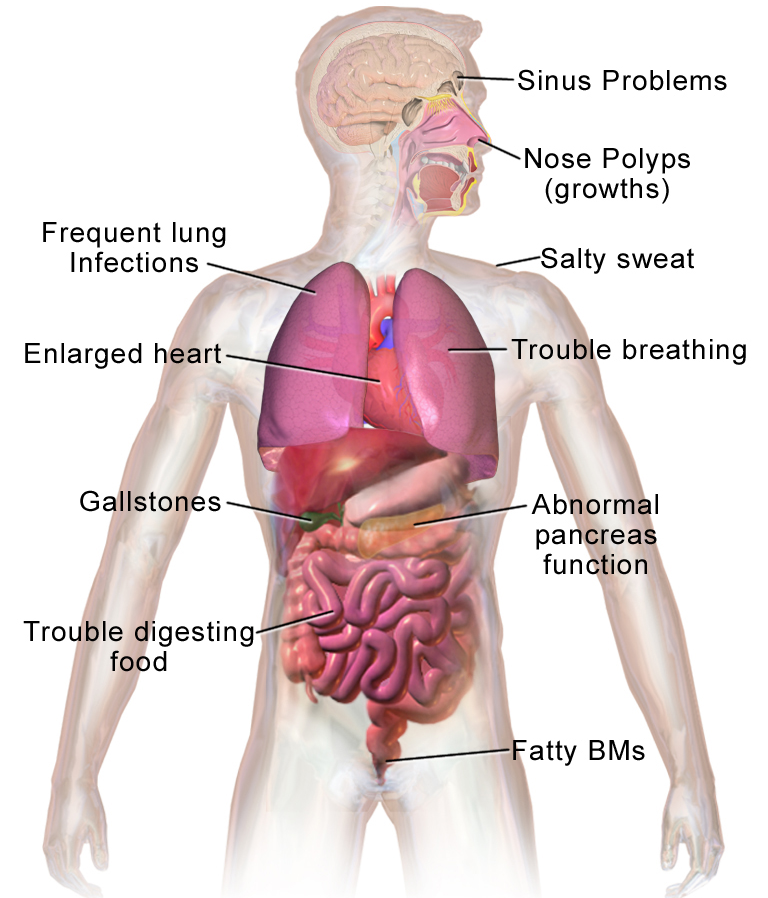
CF hinders the release of digestive enzymes from the pancreas, which triggers malnutrition, causes lung disease that is eventually fatal and produces high levels of salt in sweat that can be life-threatening.

{kind=link}
In recent years, scientists have revealed many aspects of this deadly lung disease which have led to routine early diagnosis in screened babies, better treatments and longer lives. On the other hand, the scientific community hasn’t been able to figure out when, where and why the mutation became so common. Collaborating with an extraordinary team of European scientists such as David Barton in Ireland and Milan Macek in the Czech Republic, in particular a group of brilliant geneticists in Brest, France led by Emmanuelle Génin and Claude Férec, we believe that we now know where and when the original mutation arose and in which ancient tribe of people.
We share these findings in an article in the European Journal of Human Genetics which represents the culmination of 20 years’ work involving nine countries.
What is cystic fibrosis?
My quest to determine how CF arose and why it’s so common began soon after scientists discovered the CFTR gene causing the disease in 1989. The most common mutation of that gene that causes the disease was called F508del. Two copies of the mutation – one inherited from the mother and the other from the father – caused the lethal disease. But, inheriting just a single copy caused no symptoms, and made the person a “carrier.”
I had been employed at the University of Wisconsin since 1977 as a physician-scientist focusing on the early diagnosis of CF through newborn screening. Before the gene discovery, we identified babies at high risk for CF using a blood test that measured levels of protein called immunoreactive trypsinogen (IRT). High levels of IRT suggested the baby had CF. When I learned of the gene discovery, I was convinced that it would be a game-changer for both screening test development and epidemiological research.
That’s because with the gene we could offer parents a more informative test. We could tell them not just whether their child had CF, but also whether they carried two copies of a CFTR mutation, which caused disease, or just one copy which made them a carrier.

{kind=link}
One might ask what is the connection between studying CF newborn screening and learning about the disease origin. The answer lies in how our research team in Wisconsin transformed a biochemical screening test using the IRT marker to a two-tiered method called IRT/DNA.
Because about 90 percent of CF patients in the U.S. and Europe have at least one F508del mutation, we began analyzing newborn blood for its presence whenever the IRT level was high. But when this two-step IRT/DNA screening is done, not only are patients with the disease diagnosed but also tenfold more infants who are genetic carriers of the disease are identified.
As preconception-, prenatal- and neonatal screening for CF have proliferated during the past two decades, the many thousands of individuals who discovered they were F508del carriers and their concerned parents often raised questions about the origin and significance of carrying this mutation themselves or in their children. Would they suffer with one copy? Was there a health benefit? It has been frustrating for a pediatrician specializing in CF to have no answer for them.
The challenge of finding origin of the CF mutation
I wanted to zero in on when this genetic mutation first starting appearing. Pinpointing this period would allow us to understand how it could have evolved to provide a benefit – at least initially – to those people in Europe who had it. To expand my research, I decided to take a sabbatical and train in epidemiology while taking courses in 1993 at the London School of Hygiene and Tropical Medicine.
The timing was perfect because the field of ancient DNA research was starting to blossom. New breakthrough techniques like the Polymerase Chain Reaction made it possible to study the DNA of mummies and other human archaeological specimens from prehistoric burials. For example, early studies were performed on the DNA from the 5,000-year-old Tyrolean Iceman, which later became known as Ötzi.

I decided that we might be able to discover the origin of CF by analyzing the DNA in the teeth of Iron Age people buried between 700-100 B.C. in cemeteries throughout Europe.
Using this strategy, I teamed up with archaeologists and anthropologists such as Maria Teschler-Nicola at the Natural History Museum in Vienna, who provided access to 32 skeletons buried around 350 B.C. near Vienna. Geneticists in France collected DNA from the ancient molars and analyzed the DNA. To our surprise, we discovered the presence of the F508del mutation in DNA from three of 32 skeletons.

This discovery of F508del in Central European Iron Age burials radiocarbon-dated to 350 B.C. suggested to us that the original CF mutation may have arisen earlier. But obtaining Bronze Age and Neolithic specimens for such direct studies proved difficult because fewer burials are available, skeletons are not as well-preserved and each cemetery merely represents a tribe or village. So rather than depend on ancient DNA, we shifted our strategy to examine the genes of modern humans to figure out when this mutation first arose.
Why would a harmful mutation spread?
To find the origin of CF in modern patients, we knew we needed to learn more about the signature mutation – F508del – in people who are carriers or have the disease.
This tiny mutation causes loss of one amino acid out of the 1,480 amino acid chain and changes the shape of a protein on the surface of the cell that moves chloride in and out of the cell. When this protein is mutated, people carrying two copies of it – one from the mother and one from the father – are plagued with thick sticky mucus in their lungs, pancreas and other organs. The mucus in their lungs allows bacteria to thrive, destroying the tissue and eventually causing the lungs to fail. In the pancreas, the thick secretions prevent the gland from delivering the enzymes the body needs to digest food.
So why would such a harmful mutation continue to be transmitted from generation to generation?
A mutation as harmful as F508del would never have survived among people with two copies of the mutated CFTR gene because they likely died soon after birth. On the other hand, those with one mutation may have a survival advantage, as predicted in Darwin’s “survival of the fittest” theory.
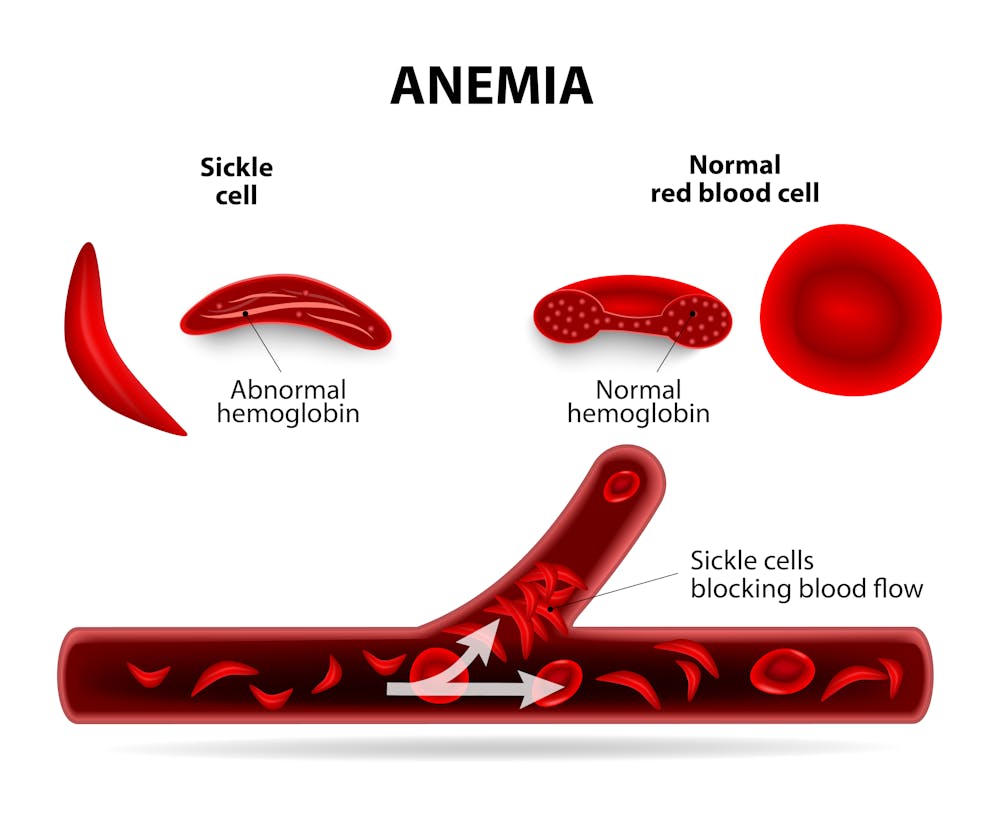
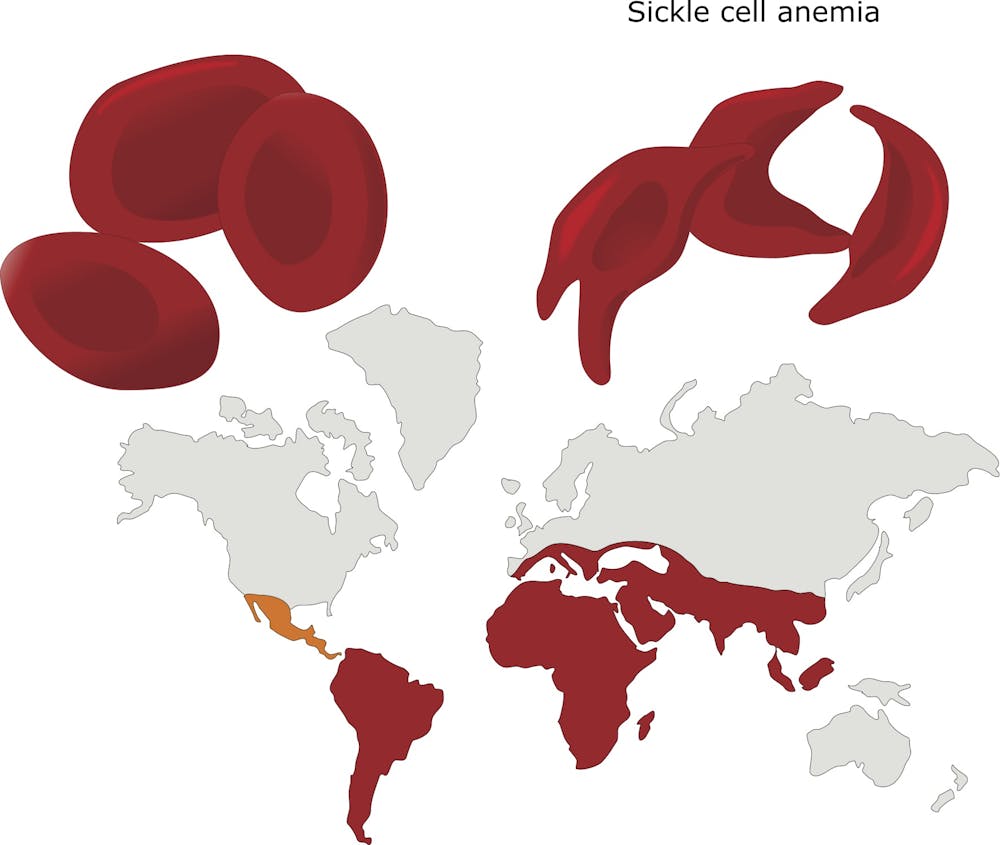
Perhaps the best example of a mutation favoring survival under stressful environmental conditions can be found in Africa, where fatal malaria has been endemic for centuries. The parasite that causes malaria infects the red blood cells in which the major constituent is the oxygen-carrying protein hemoglobin. Individuals who carry the normal hemoglobin gene are vulnerable to this mosquito-borne disease. But those who are carriers of the mutated “hemoglobin S” gene, with only one copy, are protected from severe malaria. However two copies of the hemoglobin S gene causes sickle cell disease, which can be fatal.
Here there is a clear advantage to carrying one mutant gene – in fact, about one in 10 Africans carries a single copy. Thus, for many centuries an environmental factor has favored the survival of individuals carrying a single copy of the sickle hemoglobin mutation.
Similarly we wondered whether there was a health benefit to carrying a single copy of this specific CF mutation during exposures to environmentally stressful conditions. Perhaps, we reasoned, that’s why the F508del mutation was common among Caucasian Europeans and Europe-derived populations.
Clues from modern DNA
To figure out the advantage of transmitting a single mutated F508del gene from generation to generation, we first had to determine when and where the mutation arose so that we could uncover the benefit this mutation conferred.
We obtained DNA samples from 190 CF patients bearing F508del and their parents residing in geographically distinct European populations from Ireland to Greece plus a Germany-derived population in the U.S. We then identified a collection of genetic markers – essentially sequences of DNA – within the CF gene and flanking locations on the chromosome. By identifying when these mutations emerged in the populations we studied, we were able to estimate the age of the most recent common ancestor.
Next, by rigorous computer analyses, we estimated the age of the CF mutation in each population residing in the various countries.
We then determined that the age of the oldest common ancestor is between 4,600 and 4,725 years and arose in southwestern Europe, probably in settlements along the Atlantic Ocean and perhaps in the region of France or Portugal. We believe that the mutation spread quickly from there to Britain and Ireland, and then later to central and southeastern European populations such as Greece, where F508del was introduced only about 1,000 years ago.
Who spread the CF mutation throughout Europe?
Thus, our newly published data suggest that the F508del mutation arose in the early Bronze Age and spread from west to southeast Europe during ancient migrations.
Moreover, taking the archaeological record into account, our results allow us to introduce a novel concept by suggesting that a population known as the Bell Beaker folk were the probable migrating population responsible for the early dissemination of F508del in prehistoric Europe. They appeared at the transition from the Late Neolithic period, around 4000 B.C., to the Early Bronze Age during the third millennium B.C. somewhere in Western Europe. They were distinguished by their ceramic beakers, pioneering copper and bronze metallurgy north of the Alps and great mobility. All studies, in fact, show that they were into heavy migration, traveling all over Western Europe.
{kind=link}
Over approximately 1,000 years, a network of small families and/or elite tribes spread their culture from west to east into regions that correspond closely to the present-day European Union, where the highest incidence of CF is found. Their migrations are linked to the advent of Western and Central European metallurgy, as they manufactured and traded metal goods, especially weapons, while traveling over long distances. It is also speculated that their travels were motivated by establishing marriage networks. Most relevant to our study is evidence that they migrated in a direction and over a time period that fit well with our results. Recent genomic data suggest that both migration and cultural transmission played a major role in diffusion of the “Beaker Complex” and led to a “profound demographic transformation” of Britain and elsewhere after 2400 B.C.

{kind=link}
Determining when F508del was first introduced in Europe and discovering where it arose should provide new insights about the high prevalence of carriers – and whether the mutation confers an evolutionary advantage. For instance, Bronze Age Europeans, while migrating extensively, were apparently spared from exposure to endemic infectious diseases or epidemics; thus, protection from an infectious disease, as in the sickle cell mutation, through this genetic mutation seems unlikely.
As more information on Bronze Age people and their practices during migrations become available through archaeological and genomics research, more clues about environmental factors that favored people who had this gene variant should emerge. Then, we may be able to answer questions from patients and parents about why they have a CFTR mutation in their family and what advantage this endows.