

Parallèlement à la modélisation du comportement et de l’évolution de la pandémie de Covid-19 à l’échelle macroscopique, les techniques de simulation numérique permettent d'étudier l’évolution microscopique du virus et les mécanismes responsables de l’infection humaine ou de leur contagiosité à l’échelle des atomes et des molécules. La connaissance des phénomènes mis en jeu à cette échelle apporte des éléments fondamentaux pour le développement de médicaments ou de vaccins.
Compte tenu de l’urgence sanitaire, nous avons réuni un consortium international afin d’analyser au niveau moléculaire le mode d’infection virale des coronavirus.
Les protéines du virus changent de forme quand il agit sur nos cellules
Qu’est-ce que la simulation moléculaire ? Cette discipline vise à appliquer des algorithmes basés sur des lois physiques, par exemple les équations de Newton ou les lois de la physique quantique, pour décrire le mouvement relatif de tous les atomes composant notre système d’intérêt, par exemple des protéines ou des acides nucléiques interagissant entre eux.
Dans un certain sens, il s’agit de construire un microscope ultime, virtuel, qui nous permet de voir chaque atome, ou même chaque électron. Notre « microscope » montre comment les atomes évoluent, et donc comment la structure biologique du virus change de forme au cours du temps. Ce type d’analyse permet l’élaboration d’une vidéo représentant comment le coronavirus infecte une cellule humaine ou comment il échappe à la reconnaissance du système immunitaire.
Bien évidement pour que le film soit le plus conforme possible à la réalité, il faut que les approximations faites pour définir le modèle soient bien maîtrisées et les plus précises possible. La réalisation de ce type de film est particulièrement coûteuse en termes de temps et de ressources de calcul : pour décrire une échelle de temps de quelques centaines de nanosecondes, il faudrait plusieurs mois de calculs, voire des années. C’est pourquoi l’utilisation de supercalculateurs, ici le supercalculateur Jean Zay, basés sur des architectures massivement parallèles, est fondamentale pour obtenir des résultats statistiquement significatifs en des temps raisonnables.
Malgré cela, aujourd’hui la simulation moléculaire permet d’apporter des réponses originales à de nombreuses questions biologiques, par exemple l’influence des dommages à l’ADN dans le développement des cancers, ou expliquer les mécanismes qui induisent une hypersensibilité visuelle dans des conditions de basse luminosité qui représente un effet collatéral de certains médicaments contre le cancer.
La simulation moléculaire doit absolument être utilisée de concert avec des techniques expérimentales afin d’apporter l’ensemble des éléments permettant de mieux comprendre les processus pathologiques.
Comment le SARS-CoV-2 infecte les cellules humaines
Nous avons simulé le comportement et les mécanismes d’action de deux protéines virales importantes dans le SARS-CoV-2 : la protéine dite « spike », et le domaine unique « SARS Unique Domain », ou SUD. La protéine spike est une protéine de l’enveloppe virale. Elle lui confère sa forme typique de « couronne » et permet l’entrée du virus dans les cellules humaines.
La protéine SUD se trouve dans l’enveloppe du virus et peut permettre au virus d’échapper à la surveillance du système immunitaire en interagissant avec des brins d’ARN de la cellule hôte ou infectée. Ces deux protéines peuvent donc être à l’origine de la dangerosité et de la contagiosité de ce nouveau coronavirus.
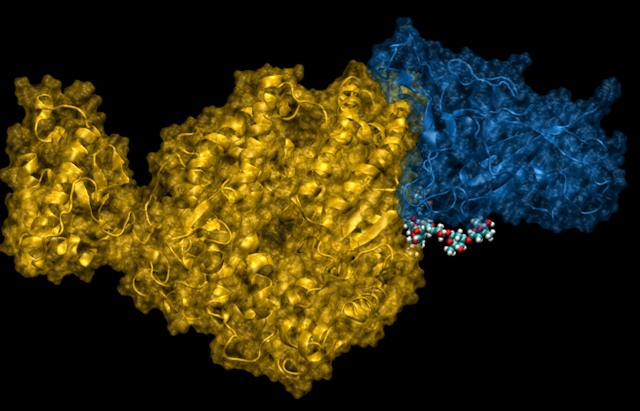
La protéine spike se lie à la surface des cellules, spécialement dans les poumons. Côté cellules humaines, il y a un « récepteur », dit ACE2 ; côté protéine spike, il y a une région dite « domaine d’accrochage au récepteur », ou « RBD ».

.png){kind=link}
L’ancrage entre récepteur humain et domaine d’accrochage du virus est la première étape du processus qui amène à la fusion de la membrane virale avec celle de la cellule humaine, et donc à l’infection. La forte affinité de SARS-CoV-2 pour le récepteur ACE2 est une des raisons de sa grande contagiosité, mais aussi des complications respiratoires engendrées par cette infection et de l’augmentation de la dangerosité pour des patients atteints d’hypertension.
Nos simulations moléculaires décryptent la formation d’un complexe stable entre RBD et ACE2, en identifiant les points d’ancrage les plus importants et les mécanismes sous-jacents.
Tester virtuellement l’action de nouveaux médicaments
Ces résultats permettent de comprendre la première étape de l’infection par le SARS-CoV-2, mais aussi de tester, toujours à l’aide de la simulation moléculaire, l’influence de certains médicaments ou composés naturels sur cette interaction. De manière intéressante, certains composés testés permettaient d’entraîner une forte déstabilisation du complexe entre RBD et ACE2, comme montré par l’augmentation de la distance entre les deux protéines en présence du médicament.
Ces résultats prometteurs devront être confirmés par des tests in vitro et in vivo pour conclure quant à leur efficacité réelle, mais ils permettent d’apporter une piste intéressante pour le développement ou le repositionnement de médicaments contre le SARS-CoV-2.
Comment le SARS-CoV-2 s’arrime sur l’ARN de nos cellules et l’empêche de faire son travail
La protéine SUD permet au virus de se camoufler et de ne pas être reconnu comme pathogène au sein des cellules infectées, ce qui explique son importante capacité d’infection.
Notre hypothèse est que la protéine SUD du virus pourrait séquestrer des brins d’ARN messager de la cellule cible, ce qui empêcherait la production de protéines spécifiques par la cellule. Normalement, le travail de ces protéines est d'amener à la destruction de la cellule avant la maturation du virus, ce qui met fin à la transmission virale et freine la propagation de l’infection. D’un point de vue structural, la protéine SUD est formée de deux sous-unités qui doivent être refermées sur elles-mêmes pour exercer son action.

Nous avons montré que la forme « compacte », ou fermée, est maintenue grâce à une interaction avec l’ARN des cellules cibles. En effet, il existe dans les cellules des arrangements particuliers des acides nucléiques qui composent l’ADN et l’ARN, appelés quadruplexes de guanine. Ces arrangements modifient la structure canonique de la double hélice puisqu’ils forment des empilements de forme carrée où quatre bases nucléiques sont reliées entre elles via des liaisons hydrogènes.
Nos simulations moléculaires montrent que la protéine SUD peut s’arrimer efficacement à l’ARN en forme de quadruplexes de guanine. Il y a deux modes d’arrimage : le premier mode permettrait de « reconnaître » des quadruplexes de guanine le long de l’ARN cible, avant de se fermer pour les incapaciter. Le second mode, quant à lui, permettrait de stabiliser la forme compacte de la protéine SUD sur l’ARN et donc d’empêcher l’élimination des cellules infectées par le système immunitaire.
Ici aussi, les séquences impliquées dans les interactions entre ARN et protéine SUD pourront être visées spécifiquement pour le développement rationnel de nouveaux médicaments.
Méthode scientifique et lutte contre la Covid-19
La résolution à l’échelle des atomes et des molécules pourra aussi permettre d’étudier comment certaines mutations du virus pourraient affecter les phénomènes étudiés, et donc de prévenir ou de contrer les effets dus à de potentiels mutants.
Bien évidemment, comme toute méthode scientifique, les simulations moléculaires ont aussi leurs limites, basées essentiellement sur la précision du modèle utilisé, mais aussi sur l’extension de l’échantillonnage statistique. Par exemple, d’autres modes d’ancrage pour les protéines ou les médicaments visés ne peuvent pas être totalement exclus et pourraient apparaître en prolongeant la durée de nos simulations. Il ne faut donc pas tirer de conclusions hâtives à partir d’une seule expérience, au contraire la comparaison avec des données issues de techniques complémentaires est toujours fondamentale. Ceci n’est, au fond, rien d’autre que la base de la méthode scientifique, qui, comme disait Karl Popper, doit se baser sur le contrôle et la réfutabilité des hypothèses et des résultats. Abandonner ces principes, spécialement dans un contexte de profonde crise sanitaire et social est fortement irresponsable et ne saurait pas être cautionné.
Ces travaux ont été possible par l’engagement fort du CNRS et en particulier des centres de calculs nationaux GENCI et IDRIS, qui ont donné un accès prioritaire au supercalculateur Jean Zay, un de plus puissant à l’échelle mondiale, pour la réalisation de nos simulations. Nos résultats montrent aussi la force et la nécessité de la collaboration internationale : notre consortium international réunit différentes équipes de l’Université de Lorraine et du CNRS, de l’Université d’Alcalá de Henares et Valencia en Espagne et de l’Université de Palerme en Italie.