

When you buy a diet coke, or any other consumables containing the artificial sweetener aspartame, you’ll see a warning against consuming the product if you have phenylketonuria, an inherited metabolic disorder.
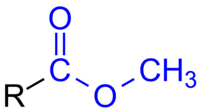
“Artificial” sweeteners, such as NutraSweet and Equal, are not saccharides – the simple carbohydrates we call sugars. Instead, the sweetener aspartame is a methyl ester comprising two joined amino acids: aspartic acid and phenylalanine (Phe) – this is important for later so keep it in mind.
{kind=link}
The safety of aspartame for general consumption has attracted attention since its discovery in 1965, but there’s no evidence of association with adverse effects. The warning on products containing aspartame is specifically for those with the severe disorder known as phenylketonuria.
Genetic basis
For someone to have phenylketonuria, they have to inherit an incorrect copy of a gene involved in Phe breakdown from each parent. Typically, the parents of an affected person (known as carriers) each have one correct and one incorrect copy of the gene, so are unaffected themselves.
The main feature of the condition is the body’s inability to break down excess Phe (recall that this one of the two amino acids in aspartame). Now, this part gets complicated but stay with me.
Amino acids can join linearly and fold into three dimensional shapes to construct functional proteins that perform a wide range of roles in our bodies. We produce some amino acids ourselves, but others, including Phe, must be obtained directly from our diet.
Amino acids can’t be stored by the body if we consume them in excess (unlike fats, for instance, and carbohydrates), so we need a constant supply. They also can’t be directly eliminated from the body, which means when excess amino acids are ingested, each type has to be broken down in a specific sequence of steps before excretion.
The first step in the degradation of Phe is its conversion to tyrosine, an amino acid important in neurotransmission (when signalling molecules from one neuron bind to and activate another).
This reaction requires an enzyme as well as the assistance of a sidekick (enzyme co-factor). The enzyme is produced through expression of its corresponding gene, and a problem arises when there are mutations in this gene.
There are 548 separate mutations recorded for this gene that lead to the production of a differing enzyme, which means the body may not be able to break Phe down.
This differing enzyme has a reduced ability of perform the initial step in the breakdown of Phe, so the level of the amino acid in the body rises. And this has a toxic effect on neurons. Early detection of high Phe levels and intervention is vital to avoid severe mental disability.
Diagnosis and treatment
Phenylketonuria was first identified by Asbjorn Folling, a biochemist and physician, who noticed an unusual odour in the urine of some individuals with developmental delays. This smell was due to a molecule produced by the body when Phe accumulates.
A diagnostic test that worked reliably from around eight weeks of age (a drop of ferric copper added to a wet nappy would turn green in a positive test) was developed in the late 1950s. The problem was that, by this age, untreated babies had often already suffered brain damage.
Only a few years later, a new, more sensitive method permitted detection from three days after birth. The Guthrie or heel-prick test requires only a drop of blood from an infant. This blood is spotted on a paper disk and placed on growth media featuring bacteria unable to synthesise Phe.
Growing bacteria – because the Phe present in the blood spot supplements what they are unable to produce themselves – represents a positive result.
The heel-prick test is widely performed as part of neonatal screening programs. But the diagnostic test now uses a technique called tandem mass spectrometry and screening includes a wide range of conditions.
In the 1950s, a low-Phe diet was introduced for people with this severe metabolic disorder and it continues to be the predominant treatment. Since Phe is found in most food sources, the diet involves getting most energy intake from a formula instead of meals, supplemented by a small amount of foods low in protein (such as fruit and vegetables).
The restrictive nature of this diet means researchers are still looking for better treatments avenues. A synthetic form of the enzyme co-factor is one option, as it an enzyme able to break down Phe and gene therapy.
But the reason why people with phenylketonuria can’t have aspartame-sweetened food is because, during digestion, it can separate into its component amino acids (aspartic acid and Phe). And this is bad news for people with the disorder.