
Cada dos años se celebra una competición para dirimir cuál es el software que predice mejor la estructura de proteínas a partir de sus secuencias de aminoácidos. El concurso se conoce por las siglas CASP (del inglés Critical Assessment of Protein Structure Prediction). Este año se ha celebrado la decimocuarta edición de la competición (CASP14), y ha resultado vencedor el algoritmo AlphaFold 2, desarrollado por la empresa DeepMind, con una gran ventaja sobre sus competidores.
Los resultados obtenidos por AlphaFold 2 han sido calificados de espectaculares por los especialistas por el alto nivel de acierto de las predicciones. Pero vayamos por partes y, para empezar, expliquemos algunas nociones básicas relativas a la estructura de las proteínas y su importancia.
Asociamos las proteínas a la musculatura de los animales, incluida la nuestra. Es lógico, dado el alto contenido que tiene el tejido muscular. Son filamentos de esas moléculas las estructuras cuyo deslizamiento genera la contracción. Pero esa no es su única función, ya que cumplen multitud de tareas en todos los organismos: actúan como si fuesen máquinas que desempeñan muy diferentes funciones.
Además de ser las responsables de que se contraigan los músculos y, de esa forma, trabajen, las proteínas cumplen un buen número de cometidos de gran importancia. Catalizan casi todas las reacciones químicas que tienen lugar en los organismos. Llevan oxígeno de los órganos respiratorios a los tejidos. Transportan (o ayudan a hacerlo) sustancias del exterior al interior de las células. Configuran arquitecturas celulares internas que cumplen funciones variadas. Reciben señales del exterior de la célula y transfieren la información al interior. Y muchas otras tareas.
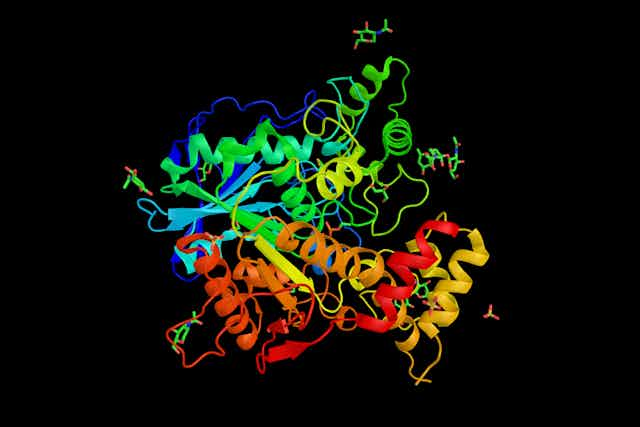
Las proteínas son cadenas de longitudes diferentes cuyos eslabones son aminoácidos, pequeñas moléculas formadas por carbono, hidrógeno, oxígeno y nitrógeno, y uno de ellos, la cisteína, también azufre. La inmensa mayoría de seres vivos no tiene más de veinte de esas moléculas. La estructura tridimensional de las proteínas depende de su composición, o sea, de los aminoácidos que las constituyen y del orden preciso en que se disponen en la cadena. Esa estructura es muy importante, porque determina su función, y es muy delicada, por lo que factores tales como la radiación, el calor y el pH la pueden alterar, lo que impide que la proteína la desempeñe o que lo haga de forma correcta.
En la actualidad, para conocer la estructura de las proteínas se utilizan varios métodos. El más tradicional es la cristalografía de rayos X, basada en el análisis del patrón de difracción que se forma cuando estos se dirigen a una sustancia en estado cristalino. En esta técnica, la interacción de los rayos X con la nube de electrones del cristal genera una imagen característica, el patrón de difracción, que permite deducir la posición de los átomos y, por lo tanto, la estructura de la molécula. El problema es que este método es laborioso y no es aplicable a muchas estructuras.
Más reciente es la criomicroscopía electrónica, una modalidad de microscopía que trabaja con muestras congeladas a temperaturas bajísimas, de manera que se evita la aparición de artefactos.
Comparar mediante aprendizaje profundo
La mayoría de las predicciones de AlphaFold 2 arrojan resultados muy similares a los que se obtienen a partir de las observaciones mediante cristalografía de rayos X, sin descartar que las predicciones mejoren, incluso, los resultados de las observaciones experimentales de algunas proteínas.
AlphaFold no deduce la estructura tridimensional de las proteínas a partir de sus propiedades fisicoquímicas. Lo que hace es comparar, mediante aprendizaje profundo haciendo uso de una red neuronal artificial, estructuras y secuencias de aminoácidos para las ciento setenta mil proteínas para las que se cuenta con el conocimiento necesario (de los doscientos millones que existen en la naturaleza). Y a partir de esa comparación es capaz de predecir la forma de proteínas cuya estructura se desconoce, pero de las que se sabe su secuencia.
Los creadores de AlphaFold sostienen que este desarrollo es la puerta que abrirá el paso al diseño y producción de fármacos con la forma adecuada para actuar sobre dianas específicas, así como a otros avances en áreas de medicina y biología en las que es importante conocer la estructura de las proteínas que participan en procesos de interés. Quizás esas pretensiones sean prematuras, pero el logro es impresionante, y marcará un antes y un después en el conocimiento de las estructuras de los seres vivos y en sus posibles aplicaciones.