
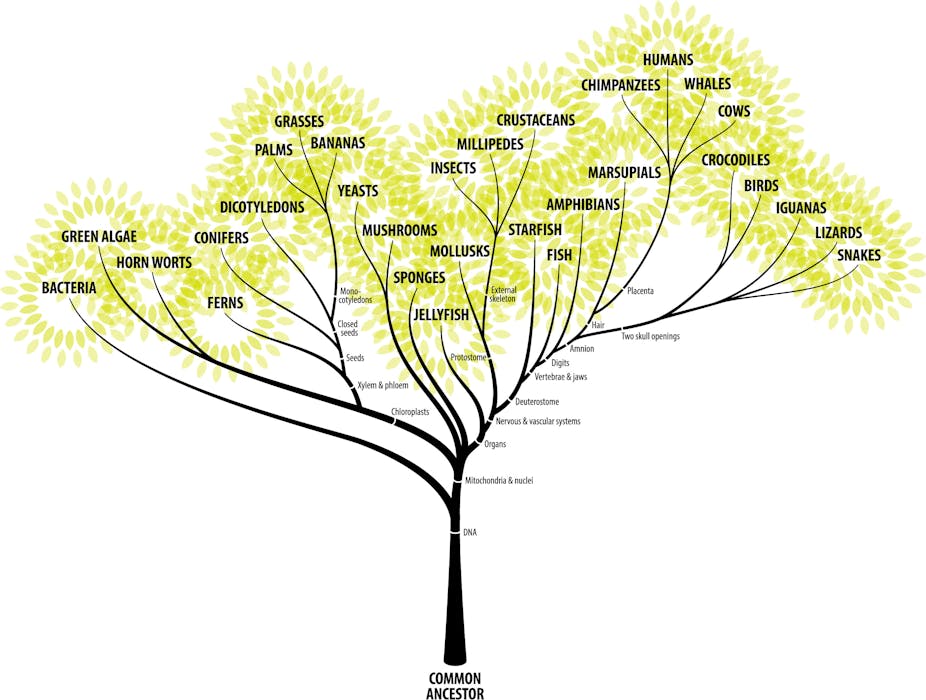
You’ve seen them in popular science news, biology textbooks, wall plaques in museums, perhaps even as tattoos. Evolutionary trees are among the most instantly recognisable, ubiquitous and iconic images of science.
One of my personal favourites – the tree of mammals published in a Science paper (you need a free account to see it) – doesn’t show all of the more than 5,000 mammals, but it contains representatives from many major groups.

I’ve simplified this tree to include common names for a selection of well-known mammals.
The resulting tree (shown above) may contain surprises for some people: for example, that humans share a more recent common ancestor with rabbits and rodents than with any other non-primate mammal; or that whales share a much more recent common ancestor with cows than dugongs.
Other research groups have produced trees of mammals that look similar, but with some species placed slightly differently.
You may wonder, then, who decides what goes where, and to what extent you should consider such trees reliable.
Some history
The idea of using a tree to represent a genealogy of species originated with the English naturalist Charles Darwin. Tree-like relationships between species had been envisaged before, but these either did not purport to represent historical descent, or lacked the crucial conceptual framework that extant species are descended from common ancestors that have since gone extinct.

{kind=link}
Most of Darwin’s tree sketches were conceptual and did not pretend to represent relationships determined from data. But empirical trees began to appear soon after the publication of his work On the Origin of Species.
These early trees were based on shared characters. For example, the trees of British biologist Saint George Jackson Mivart compared features of vertebrae and limb-bones. Species placed close in the tree share a greater number of characteristics, and are thus more similar than species placed far apart.
This approach to inferring trees is still relevant, and has been formalised as the concept of parsimony.
The best, or most parsimonious, tree is the one that assumes the smallest number of evolutionary changes. This is an application of Occam’s Razor: the conviction that the simplest explanation is preferable, all else being equal.
There are efficient algorithms for determining how many evolutionary changes are required to explain a given tree. Given any two trees, it is thus easy to determine which of them is more parsimonious.
But it’s still a difficult problem to identify the most parsimonious of all possible trees. It’s difficult because the number of trees is vast: for just 50 species, there are approximately 1076 possible trees. That’s about the same as the number of atoms in the universe!
Another problem for parsimony methods is convergent evolution. Two species may share a characteristic that their common ancestor did not have, perhaps because they have adapted to similar lifestyles or environments. Such characteristics can create a false appearance of recent common ancestry. For example, whales and dugongs have similar looking tails, but are not closely related.
Yet another problem with parsimony, identified by Mivart and still relevant today, is that a different tree may be inferred if one chooses a different set of characteristics.
Worse, considering a larger number of characteristics doesn’t necessarily make parsimony trees more accurate. Parsimony is said to lack the property of consistency.
Statistical methods
The problem of consistency can be overcome using statistical methods such as maximum likelihood and Bayesian inference. At the core of these approaches is a model of the evolutionary process. Such models associate probabilities with each possible evolutionary change.
This is most often done using molecular sequences. For example, DNA sequences contain four chemicals labelled A, C, G and T, and one can estimate the probability that an A is replaced by a G, say.
Maximum Likelihood methods calculate the probability of generating an observed set of sequences, given a tree. They then search for the tree that maximises this probability. Bayesian Inference does the opposite: it calculates the probability of the tree, given the sequences.
These methods are consistent, but this is only true if the underlying probability model is sufficiently similar to the actual evolutionary process. It is thus crucial to select an appropriate model.
Multiple lines of evidence
Perhaps the most convincing evidence that an evolutionary tree is correct is when methods based on different data infer the same, or very similar, tree.
For example, one tree of mammals was inferred by tracking transposable elements.
These are bits of DNA that insert copies of themselves into their host genome, so that those copies are inherited. Such copies can persist over vast periods of time, so that they can be used to identify common evolutionary descent of a group of species.
The tree inferred using transposable elements is remarkably similar to the tree of mammals I mentioned earlier, which was inferred by comparing DNA sequences.
Can you trust an evolutionary tree?
Any evolutionary tree should be regarded with healthy scepticism. They are working hypotheses that are likely to be revised as new evidence comes to light.
It is not possible to set aside all biases and preconceived ideas when inferring evolutionary trees, because even the methodology is based on assumptions about how evolution works.
But the better one understands the models and methods, the more one appreciates that trees are not mere guesses, nor even summaries of expert opinion.
They are products of careful and principled science informed by statistics.