
Two years ago, former President Barack Obama announced the Precision Medicine initiative in his State of the Union Address. The initiative aspired to a “new era of medicine” where disease treatments could be specifically tailored to each patient’s genetic code.
This resonated soundly in cancer medicine. Patients can already manage their cancer with therapies that target the specific genes that are altered in their particular tumor. For example, women with a type of breast cancer caused by the amplification of gene HER2 are often treated with a therapeutic called herceptin. Because these targeted therapeutics are specific to cancer cells, they tend to have fewer side effects than traditional cancer treatments with chemotherapy or radiation.
However, such treatments are not available for most cancer patients. In many cancers, the specific genetic alterations that are responsible for a cancer remain unknown. To create individualized cancer treatments, we must know more about the functional genetic alterations.
With data on cancer genetics growing rapidly, mathematics and statistics can now help unlock the hidden patterns in this data to find the genes that are responsible for an individual’s cancer. With this knowledge, physicians can select appropriate treatments that block the action of these genes to personalize therapies for individual patients. My research aims to improve precision medicine in cancer – by building on the same methods that have been used to find patterns in Netflix movie ratings.
Sifting through the data
Today, there is unprecedented public access to cancer genetics data. These data come from generous patients who donate their tumor samples for research. Scientists then apply sequencing technologies to measure the mutations and activity in each of the 20,000 genes in the human genome.
All these data are a direct result of the Human Genome Project in 2003. That project determined the sequence for all the genes that make up healthy human DNA. Since the completion of that project, the cost of sequencing the human genome has more than halved every year, surpassing the growth of computing power described in Moore’s Law. This cost reduction enables researches to collect unprecedented genetics data from cancer patients.
Most scientific studies on cancer genetics performed worldwide release their data to a centralized, public database provided by the U.S. National Institutes of Health (NIH) National Library of Medicine. The NIH National Cancer Institute and National Human Genome Research Institute have also freely released genetic data from over 11,000 tumors in 33 cancer types through a project called The Cancer Genome Atlas.
Every biological function – from extracting energy from food to healing a wound – results from activity in different combinations of genes. Cancers hijack the genes that enable people to grow to adulthood and that protect the body from the immune system. Researchers dub these the “hallmarks of cancer.” This so-called gene dysregulation enables a tumor to grow uncontrollably and form metastases in distant organs from the original tumor site.
Researchers are actively using these public data to find the set of gene alterations that are responsible for each tumor type. But this problem is not as simple is identifying a single dysregulated gene in each tumor. Hundreds, if not thousands, of the 20,000 genes in the human genome are dysregulated in cancer. The group of dysregulated genes varies in each patient’s tumor, with smaller sets of commonly reused genes enabling each cancer hallmark.
Precision medicine relies on finding the smaller groups of dysregulated genes that are responsible for biological function in each patient’s tumor. But, genes may have multiple biological functions in different contexts. Therefore, researchers must uncover a set of “overlapping” genes that have common functions in a set of cancer patients.
Linking gene status to function requires complex mathematics and immense computing power. This knowledge is essential to predict of outcome to therapies that would block the function of these genes. So, how can we uncover those overlapping features to predict individual outcomes for patients?
What Netflix can teach us
Fortunately for us, this problem has already been solved in computer science. The answer is a class of techniques called “matrix factorization” – and you’ve likely already interacted with these techniques in your everyday life.
In 2009, Netflix held a challenge to personalize movie ratings for each Netflix user. On Netflix, each user has a distinct set of ratings of different movies. While two users may have similar tastes in movies, they may vary wildly in specific genres. Therefore, you cannot rely on comparing ratings from similar users.
Instead, a matrix factorization algorithm finds movies with similar ratings among a smaller group of users. The group of users will vary for each movie. The computer associates each user with a group of movies to a different extent, based upon their individual tastes. The relationships among users are referred to as “patterns.” These patterns are learned from the data, and may find common rankings unforeseen by movie genre alone – for example, users may share a preference for a particular director or actor.
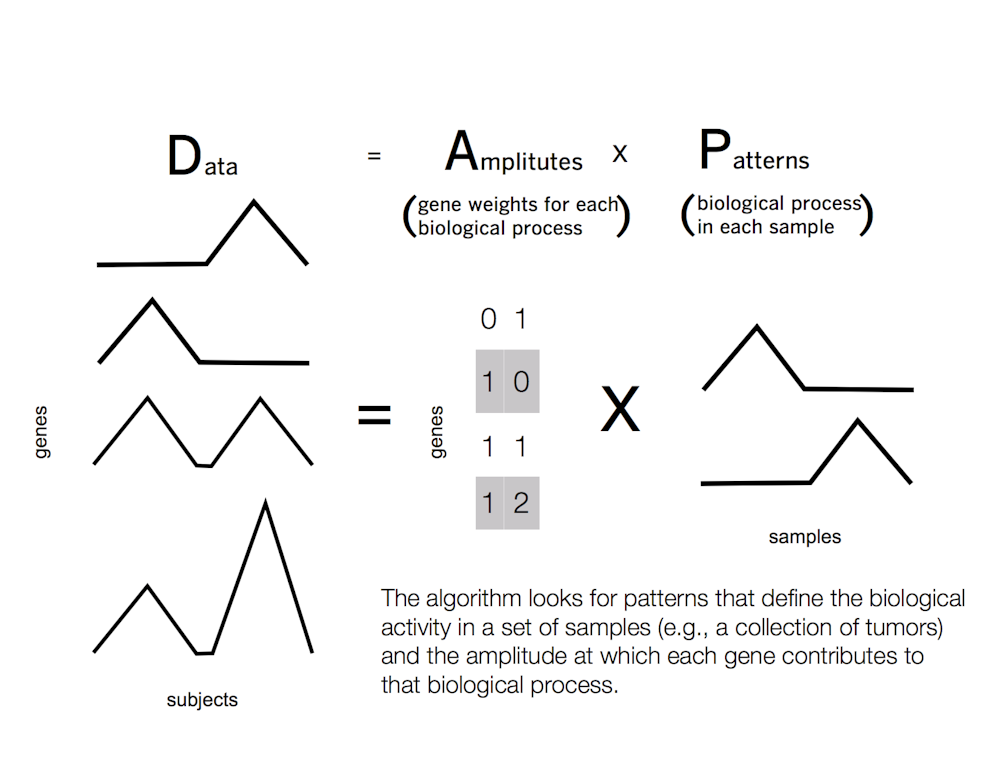
The same process can work in cancer. In this case, the measurements of gene dysregulation are analogous to movie ratings, movie genres to biological function and users to patients’ tumors. The computer searches across patient tumors to find patterns in gene dysregulation that cause the malignant biological function in each tumor.
From movies to tumors
The analogy between movie ratings and cancer genetics breaks down in the details. Unless they are minors, Netflix users are not constrained in the movies they watch. But, our bodies instead prefer to minimize the number of genes used for any single function. There are also substantial redundancies between genes. To protect a cell, one gene may easily substitute for another to serve a common function. Gene functions in cancer are even more complex. Tumors are also highly complex and rapidly evolving, depending upon random interactions between the cancer cells and the adjacent healthy organ.
To account for these complexities, we have developed a matrix factorization approach called Coordinated Gene Activity in Pattern Sets – or CoGAPS for short. Our algorithm accounts for biology’s minimalism by incorporating as few genes as possible into the patterns for each tumor.
Different genes can also substitute for one another, each serving a similar function in a different context. To account for this, CoGAPS simultaneously estimates a statistic for the so-called “patterns” of gene function. This allows us to compute the probability of each gene being used in each biological function in a tumor.
For example, many patients take a targeted therapeutic called cetuximab to prolong survival in colorectal, pancreatic, lung and oral cancers. Our recent work found that these patterns can distinguish gene function in cancer cells that respond to the targeted therapeutic agent cetuximab from those that do not.
The future
Unfortunately, cancer therapies that target genes usually cannot cure a patient’s disease. They can only delay progression for a few years. Most patients then relapse, with tumors that are no longer responsive to the treatment.
Our own recent work found that the patterns that distinguish gene function in cells that are responsive to cetuximab include the very genes that give rise to resistance. Emerging immunotherapies are promising and appear to cure some cancers. Yet, far too often, patients with these treatments also relapse. New data that track the cancer genetics after treatment is essential to determine why patients no longer respond.
Along with these data, cancer biology also requires a new generation of scientists who can bridge mathematics and statistics to determine the genetic changes occurring over time in drug resistance. In other fields of mathematics, computer programs are able to forecast long-term outcomes. These models are used commonly in weather prediction and investment strategies.
In these fields and my own previous research, we have found that updates to the models from large datasets – such as satellite data in the case of weather – improve long-term forecasts. We have all seen the effect of these updates, with weather predictions improving the closer that we are to a storm.
Just as tools from computer science used can be adapted to both movie recommendations and cancer, the future generation of computational scientists will adopt prediction tools from an array of fields for precision medicine. Ultimately, with these computational tools, we hope to predict tumors’ response to therapy as commonly as we predict the weather, and perhaps more reliably.