

Many people will never have heard of cryo-electron microscopy before the announcement that Jacques Dubochet, Joachim Frank and Richard Henderson had won the 2017 Nobel Prize in chemistry for their work developing this technology. So what is it, and why is it worthy of this honor?
Cryo-electron microscopy – or cryo-EM – is an imaging technology that allows scientists to obtain pictures of the biological “machines” that work inside our cells. Most amazingly, it can reconstruct individual snapshots into movie-like scenes that show how protein components of these biological machines move and interact with each other.
It’s like the difference between having a list of all of the individual parts of an engine versus being able to see the engine fully assembled and running. The parts list can tell you a lot, but there’s no replacement for seeing what you’re studying in action.
What’s revolutionary about cryo-EM is not only that it lets scientists actually see and understand how important biological machines work, but that it allows us to study a vast array of important proteins that can’t be seen using any other structural biology technique.
Advances in both imaging technology and computing have really pushed cryo-EM forward over the last decade or so. Researchers are now able to generate atomic, or near-atomic, resolution 3-D models of challenging molecules – things like receptors that are therapeutic drug targets, molecular motors that deliver cargo to different parts of the cell and emerging viruses that lead to human disease.
Frozen world of cryo-EM
To obtain an image using cryo-EM, researchers take proteins that have been biochemically purified from cells and instantaneously freeze the sample on a cryo-EM grid at -180 degrees Celsius. The goal of this process is to trap many copies of a single protein or a protein complex in a thin layer of vitrified ice.
This ice is transparent to the microscope’s electron beam and allows the proteins to retain their natural shape and organization. If the sample is frozen too slowly, then ice crystals form, ruining the structure of the molecules being studied and disrupting the electrons traveling through the sample.
A major advantage of this technique is that it saves time and work. Cryo-EM’s ability to look at proteins in a near-native state is in stark contrast with X-ray crystallography, the longstanding gold standard for obtaining high-resolution biomolecular images. The older technique requires the formation of ordered crystals, where the proteins must first self-organize together in repeating patterns – at best a tricky challenge, at worst an impossible one for certain molecules. With cryo-EM, there’s no need to coax biological molecules into ordered arrays.
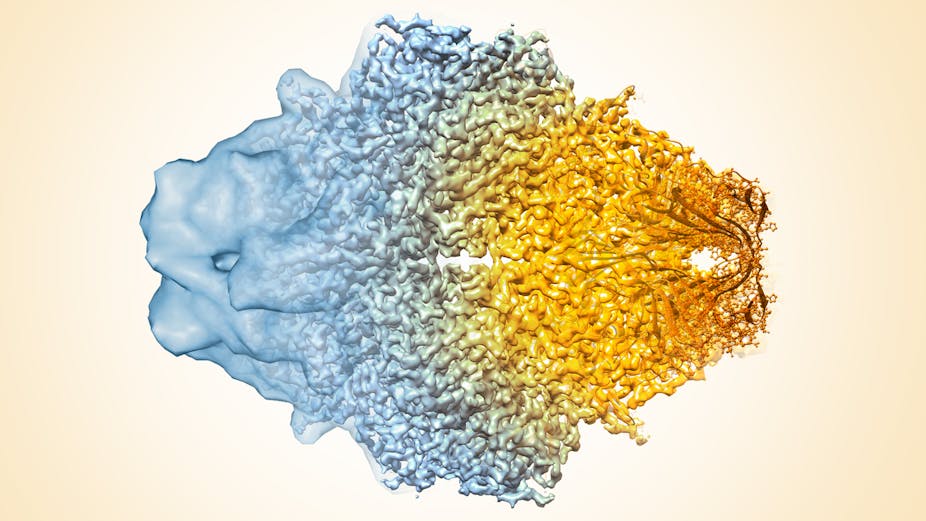
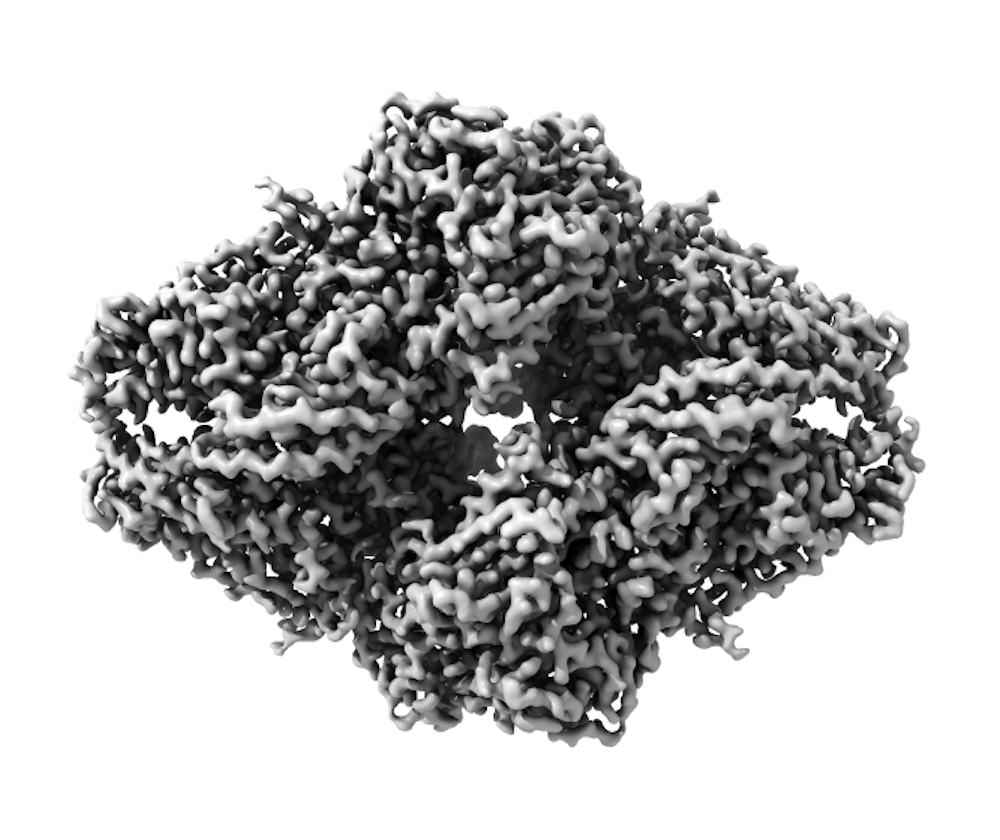
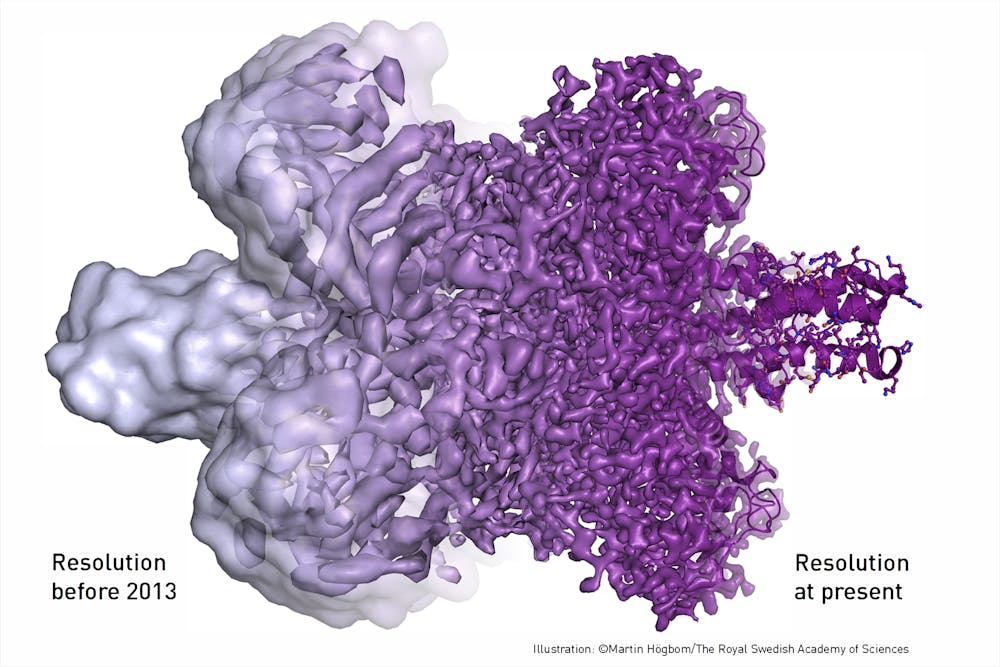
Once the cryo-EM sample has been frozen, a focused beam of electrons reveals the shape of these very small, nanometer-sized proteins. (A nanometer is about one million times smaller than the tip of a needle.) Each image contains all the information required to determine the 3-D structure. But these raw images are extremely “noisy” and hard to see, so large numbers of images for each sample must be collected using the microscope.
Specialized computer analysis then combines hundreds of thousands of individual, 2-D snapshots from different angles into a composite that can be viewed in 3-D. Many 3-D structures determined by cryo-EM are now at a high enough resolution that researchers can visualize individual atoms in the structures.

Software can also sort out proteins that are in different stages of a biological process – thus helping piece together how a biological machine moves, changes and functions. Then researchers can further test how the machine’s organization leads to specific functions in a living cell.
For example, several years ago researchers here at the University of Michigan Life Sciences Institute obtained the first 3-D snapshots of the “assembly line” within microorganisms that naturally produces a broad class of compounds known as polyketides, which includes antibiotics and other drugs. This information then gives investigators a solid blueprint for figuring out how they might redesign the microbial assembly line to produce new drugs.
One of the challenges of working with cryo-EM is that it requires massive amounts of computational power and data storage. One cryo-EM movie of 30 to 60 frames requires up to 8 gigabytes of storage – it would take only a couple of these movies to fill up most smartphones. About a thousand of these movies are collected in a day, requiring somewhere between one and eight terabytes of storage (or about 8,000 smartphones’ worth). A full data set for one 3-D structure can require 4,000 movies.
These data sets often take hundreds of thousands of hours of computer processor time to piece together. Researchers rely on supercomputers that use many processors working in parallel. The most advanced cryo-electron microscopes and accompanying computing tools require significant investments on the parts of universities, which is why they’re still relatively rare. For example, buying just the microscope and specialized camera needed to collect these cryo-EM images costs well over US$5 million.
Putting the technology to work
While the technology and technique are interesting, the true power of the tool is its ability to help us answer important biological questions.
In the Cianfrocco lab, for example, we’re using cryo-EM to look at the dynamic process of how molecular “motors” move along microtubular “tracks” inside cells. We want to figure out the basics of how cells know what to move, when. While much work has gone into identifying the necessary building blocks for moving cargo around the cell, the molecular details remain unknown.
It’s these motors that move things around the cell that aren’t working correctly in Parkinson’s, Huntington’s and Charcot-Marie-Tooth disease. Learning more about how they’re malfunctioning will be critical for developing new therapeutics for these neurodegenerative diseases. Viruses also hijack these these motor proteins during infection, which means understanding more about how they walk will help researchers design effective treatments against viruses such as HIV and rabies.

And in the Ohi lab, we’re generating detailed 3-D structures of the molecular machines bacteria use to spread disease. Biological pathogens have evolved numerous ways to infect their hosts, including toxins that alter cellular functions and complex secretion systems that inject DNA and proteins into host cells. Using 3-D snapshots of these machines, we’re hoping to find new ways to target the processes bacteria use to cause disease, such as the bacteria Helicobacter pylori’s ability to trigger chronic inflammation, which is a major risk factor for stomach cancer.
One of the larger questions for the field is how to make the specialized and expensive resources required for cryo-EM more broadly available to researchers across the country and around the world. To this end, the Cianfrocco lab is developing two separate cloud computing resources with the goal to streamline cryo-EM data processing to allow structural biologists to focus on the biology, not the hardware. By removing the computing bottlenecks, these tools have the potential to continue the growth of cryo-EM into a mainstream technique for scientists worldwide.