

Designer therapies are treatments tailored to a specific disease, and nowhere is the need greater for new therapies than in a group of nervous system disorders, known as “neurodegenerative diseases.”
Many of these diseases are common and well-known, such as Alzheimer’s or Parkinson’s disease. However, some are very rare, genetic disorders that are the consequence of a defective gene. In all these diseases, a mutant protein that misfolds causes the degeneration and death of neurons. One effective therapeutic strategy is to prevent the rogue protein from ever being made.
Spinocerebellar ataxia type 7 (SCA7) is one such disease in which nerves in different parts of the brain, including the eye, degenerate, which leads to blindness and difficulty walking, speaking and balancing. SCA7 is dominantly inherited – which means that you just need one bad copy of the mutation to cause disease. The disease occurs when a short section of DNA that encodes ataxin-7 gene is erroneously repeated – like a word in a book printed two or three times. In this case, three chemical units of the DNA sequence – C-A-G – are repeated over and over.

I became fascinated by diseases caused by these DNA repeats in 1991 when, as an M.D.-Ph.D. student, I discovered the first CAG repeat mutation responsible for a human disease called X-linked spinal and bulbar muscular atrophy, a rare neuromuscular disorder that affects only men, causing them to become weak and sometimes confined to a wheelchair. I decided to become a neurogeneticist – a geneticist who specializes in inherited neurological diseases – to focus on these disorders.
In a recent paper, my colleagues and I describe a method that blocks the production of a mutant protein to stem the progression of neurodegeneration in the eye. We hope that this strategy will be broadly applicable to halting progressive neurodegenerative disease.
How DNA repeats cause disease
Patients with SCA7 possess a string of at least 37 CAG repeats in one of their ataxin-7 genes. Because SCA7 is a dominantly inherited genetic disease, each child of an affected SCA7 patient has a 50 percent chance of inheriting the mutant gene and developing the disorder.
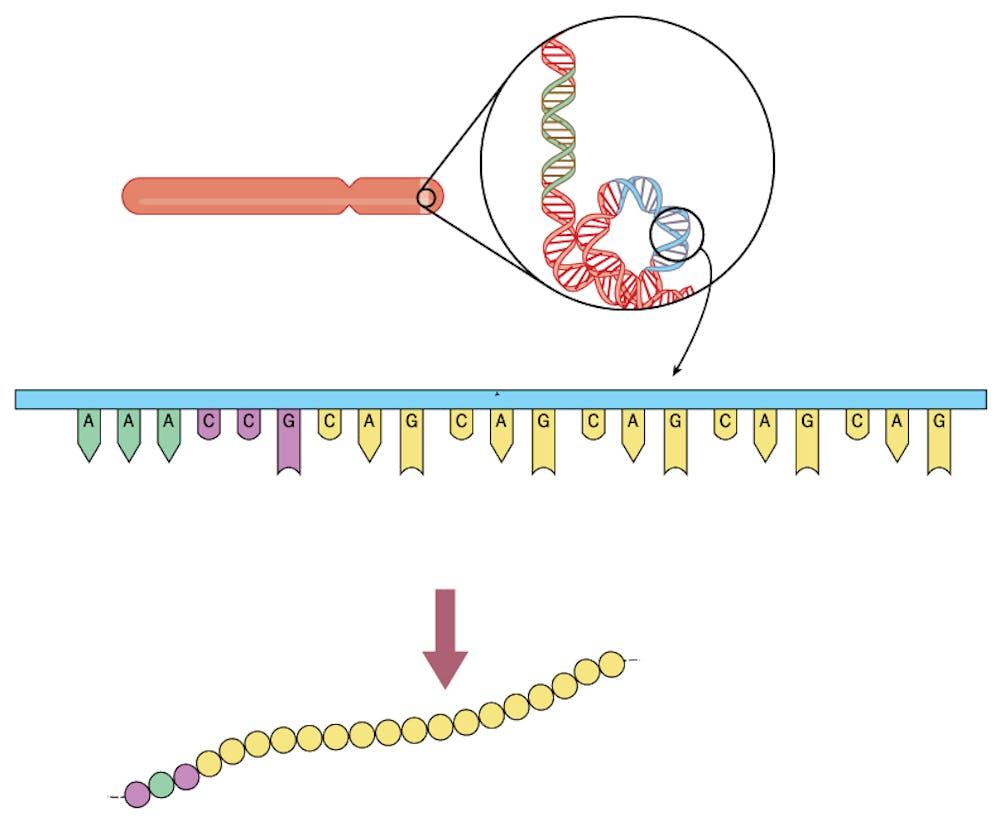
The ataxin-7 protein is part of a large collection of proteins that transcribes a gene’s DNA into a messenger RNA. When the CAG sequence extends beyond 37 repeats, it causes disease because it causes the ataxin-7 protein to “misfold.” This mutant deformed protein then accumulates in nerve cells in the brain and eye, destroying them. Coincidentally, the accumulation of a misfolded protein in SCA7 is exactly the same type of molecular problem that occurs in Alzheimer’s disease and Parkinson’s disease, where misfolded proteins, albeit different ones, accumulate in the brains of patients.
SCA7 patients suffer a double whammy when it comes to their disease. In addition to the degeneration of neurons in the cerebellum and brainstem that causes SCA7 patients to develop ataxia – loss of the ability to perform coordinated movements like walking and talking – it also causes them to go blind.
There are no drugs to treat SCA7 at this time, and the disease is relentlessly progressive, so patients succumb to the disease 10 to 25 years after onset, depending on the severity of the disease. SCA7 can trigger symptoms at any age, with many patients presenting as young adults, teenagers or even children.
My research lab and other research groups have been trying to develop a treatment for SCA7. Because the condition results from the accumulation of a toxic disease-causing protein, we reasoned that one effective therapeutic strategy would be to block the mutant protein from ever being made.
A new type of drug
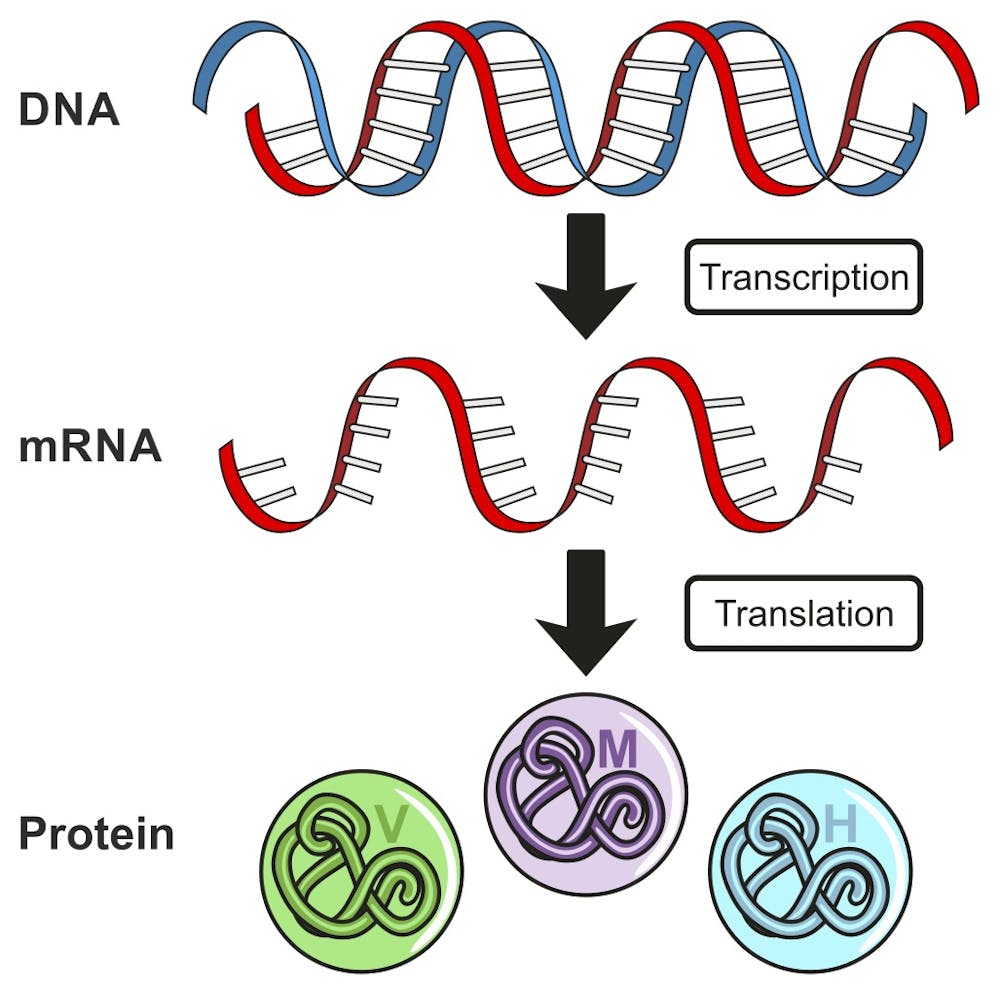
Like all genes in humans or any other organism, the ataxin-7 disease gene DNA must first be transcribed into a messenger RNA (mRNA) molecule. Then the mRNA is translated, one amino acid at a time, to produce the ataxin-7 protein.
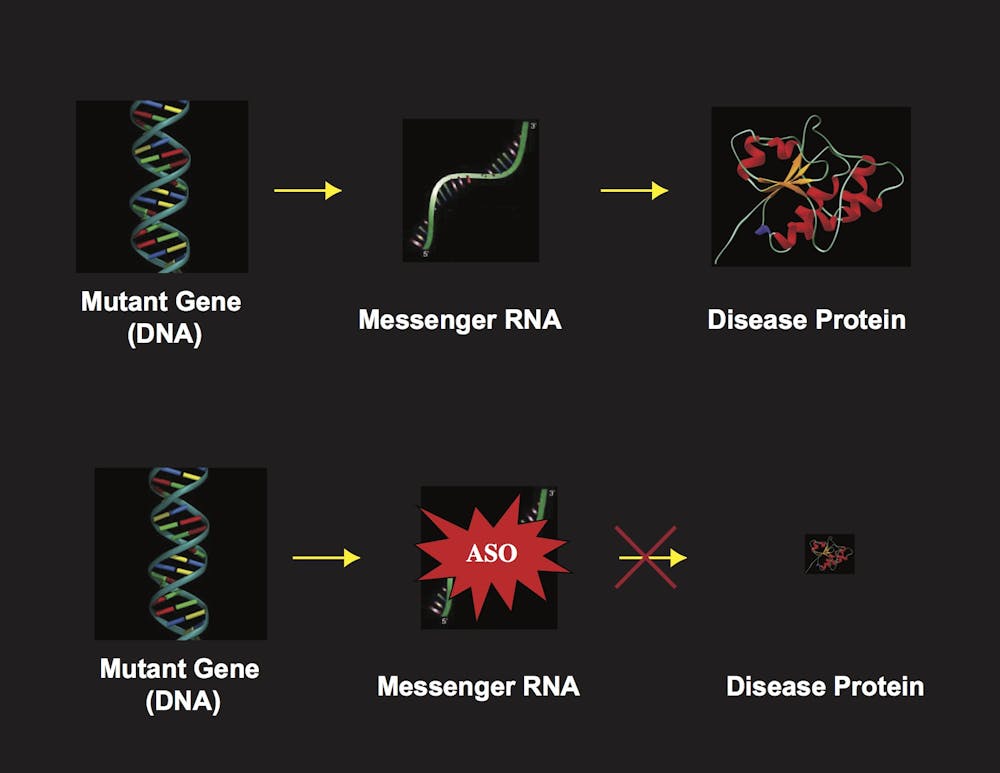
One way to prevent the disease protein from ever being produced is to destroy the mRNA before it is translated into a protein. Researchers have been working on different techniques to destroy RNAs. There are two main approaches. The one we are using, that is proving to be effective, relies upon a short segment of DNA that is synthesized in the lab and known as an antisense oligonucleotide, or ASO.
An ASO is a short stretch of DNA that is synthesized with a sequence that perfectly matches the target RNA. In the cell the ASO binds to the target mRNA and forms a duplex, a DNA-RNA hybrid molecule, which is recognized as potentially foreign and destroyed. In our latest publication, we created an ASO matching the ataxin-7 RNA, and tested whether it could treat retinal degeneration in a mouse. These mice were engineered with the human SCA7 gene mutation and were known to develop the retinal degeneration and go blind, just like their human counterparts.
We chose to focus on the eye disease first because the eye is accessible and ASOs can be delivered by injecting directly into the vitreous humor, the jelly-like substance that forms the bulk of the eyeball.
Blocking blindness
In our study, we injected an ataxin-7 ASO into one eye and a random control ASO into the other. We found that eyes injected with the ataxin-7 ASO retained the ability to see, unlike the control eyes. Because we can’t ask a mouse whether its vision is better or worse, we measured how retinal neurons respond to light stimulation, to determine whether the vision improved or deteriorated.
As expected, better vision was correlated with lower levels of ataxin-7 mRNA and less accumulation of the misfolded protein. Importantly, we also tested if delivering the ASO after onset of vision loss would prevent further deterioration of eyesight. Our results suggest our treatment might stop and even reverse blindness.
As we can perform genetic testing on individuals at risk of developing SCA7 prior to disease onset, it should be possible to initiate therapy in human SCA7 patients before symptoms appear, to prevent any eye damage. Furthermore, success in treating SCA7 eye disease suggests that using the same ASO approach to treat SCA7 brain degeneration might also work.
While the therapy that we developed is restricted to SCA7 patients, the notion of using ASOs to target RNAs that produce misfolded proteins in more common neurodegenerative diseases is well underway in labs around the globe, offering hope for patients with Huntington’s disease and amyotrophic lateral sclerosis, which is also known as Lou Gehrig’s disease.
Given the recent success with ASO therapy (Spinraza) as a treatment for infantile-onset spinal muscular atrophy, we expect that we can proceed to clinical trials in SCA7 patients, once a human-specific ataxin-7 ASO drug has been perfected and shown to be safe.
The advent of ASOs and related therapies suggest that medical research may create powerful new treatments for many neurodegenerative diseases within the next decade or two. As estimates predict that more than 20 million people will suffer from such diseases in the USA alone by 2050, these efforts are desperately needed.