

{kind=link}
The headlines are eye-catching: Scientists have sequenced the genome of white sharks. Or the bamboo lemur, or the golden eagle. But why spend so much time and money figuring out the DNA makeup of different species?
I am an evolutionary biologist at the Florida Program for Shark Research. Our research focuses on understanding how modern sharks and rays diversified over the course of their evolution to colonize the habitats they occupy today.
Rough screening of whole genomes is useful to help identify genetic markers (sequences of genes) to better understand population-level processes. But the real and enduring value of whole genome sequencing is only realized when a lot of accurate, high-resolution genomes are amassed that can be compared with one another. This type of work is just getting started.
Blueprints without instructions
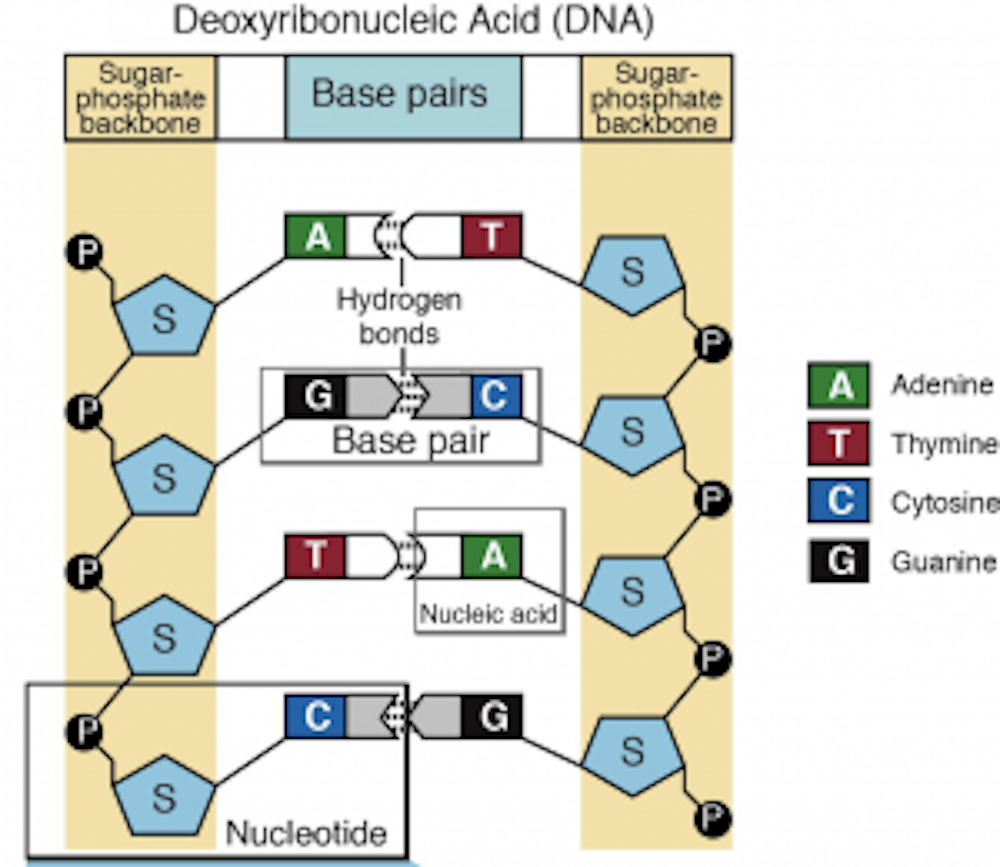
An organism’s genome – the complete catalog of its DNA – holds the blueprint for its design. Differences in the DNA sequences that make up genomes are responsible for the differences we see among individuals.
Identical twins are physically similar to one another because their genomes are identical. Siblings resemble each other because they inherit large stretches of their genomes from the same set of parents. And closely related species look more similar to each other than do those that are more distantly related, because their underlying genomes are more similar.
It follows that if we had a complete genome sequence for an organism, we would have all the information we’d need to understand how it works “from the ground up.” Indeed, this was the justification for the initial Human Genome Project
But an organism’s genomic DNA sequence can contain billions of nucleotides, or genetic building blocks. Trying to piece together what that organism might look like from its genome sequence would be like trying to make sense of thousands of concurrently transmitted telephone conversations from the “packets” of information that arrive at the receiving end of a fiber-optic telephone cable, without knowing anything about how the information was organized. The data is “all there,” but it’s hard to know what it means without an explicit interpreter. And scientists do not yet know how all of the information in genomes is organized, or how its activity is choreographed.
Learning by comparing
If it’s so hard to interpret information buried in genomes, why bother collecting the data? The answer is that if we compare genomes against one another, we can deduce which elements are responsible for particular traits.
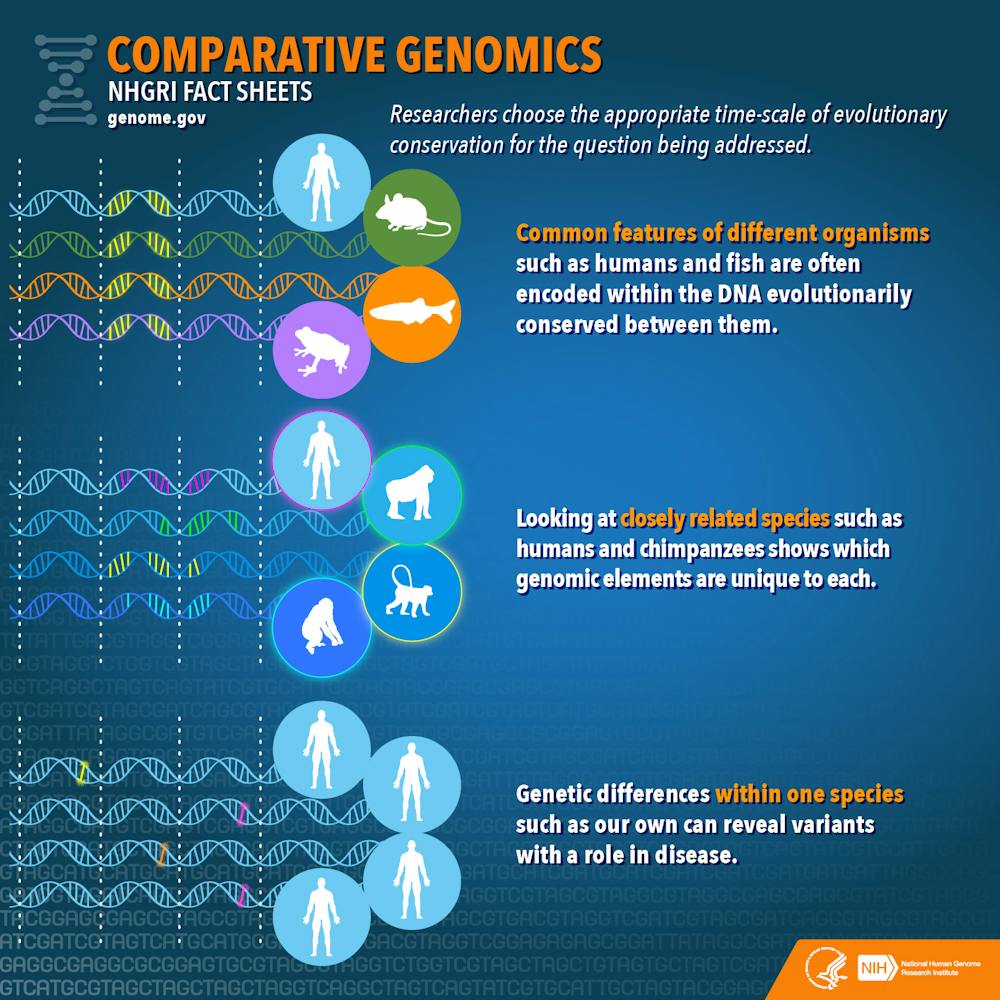
For example, humans and chimpanzees have genomes that are approximately 98 percent similar. This means that the 2 percent difference between their respective genomes must somehow account for the differences in their appearance and associated traits. Comparing the genomes side by side allows us to identify the parts of the genome responsible for the observed differences.
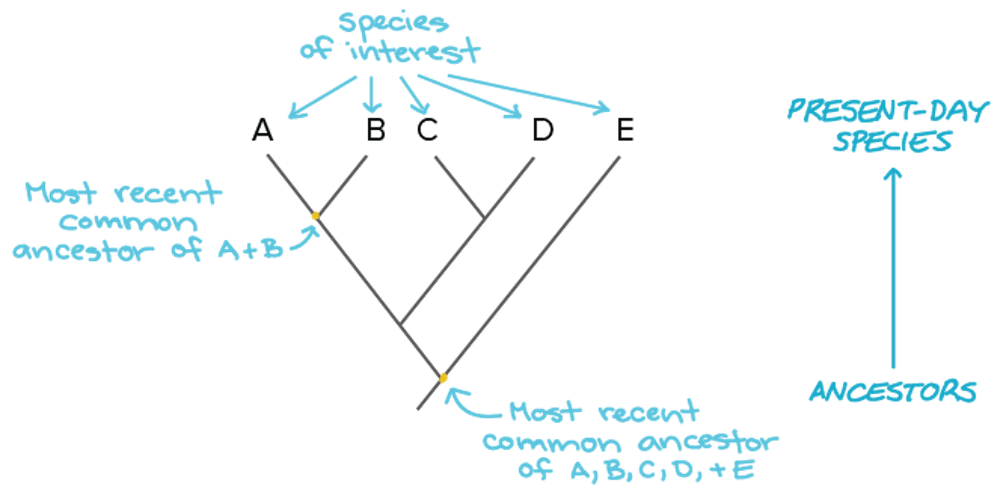
Obviously, it is important to choose carefully which comparisons to make. Comparing a human genome with a duck-billed platypus genome isn’t going to tell us much about what makes humans – or duck-billed platypuses, for that matter – so “special.” The two species diverged about 150 million years ago, and there are so many differences in their genomes and in the traits they exhibit that it would be impossible to know which genomic differences were responsible for which traits.
However, comparing human and platypus genomes (two mammals) against a bird genome would allow us to identify aspects of human and platypus genomes that were shared, but distinct from the bird genome. And in turn, comparing genomes of several mammals and birds against genomes of amphibians would help us narrow down what genomic elements birds and mammals had in common that were different from amphibians.
Building genetic libraries
Hierarchical comparisons like the one described above lie at the core of comparative genomics, a field that sets out to understand how patterns of variation in genomes are associated with, or “map to,” patterns of variation in observable traits. Biologists refer to this set of associations as the “genotype-phenotype map.”
Obviously, scientists need to know the evolutionary relationships among organisms before any of this can be done, and to make sure the genomic information we collect is accurate. If it is inaccurate or incomplete, we risk missing important associations between genotypes and the traits they code for.
Recent advances in next-generation DNA sequencing and computer science are revolutionizing the collection and analysis of this data. But it’s still expensive. It costs about US$30,000 to sequence and assemble a 2.5 billion base pair genome (for comparison, the human genome has about 3 billion base pairs) with sufficient accuracy to be useful for comparative genomic work - and more for larger genomes, such as that of the lungfish or the salamander.
An international consortium of scientists is working to collect high-quality genome sequences for all vertebrate animals that meet this standard. Initial comparisons are focusing on species selected to represent the evolutionary diversity of different groups of vertebrates – a carefully vetted set of birds, reptiles, mammals, amphibians and fishes. In September 2018 the project released its first 15 high-quality reference genomes for species including the Canadian lynx, zebra finch and blunt-snouted clingfish.
Subsequent comparisons will fill in the evolutionary gaps, until we eventually have a complete set of highly accurate genomes that can be compared with one another. These highly accurate genomes will improve our understanding of the genotype-phenotype map. They also will serve as as references for researchers trying to understand the role different genes play in guiding normal development, and for others exploring likely causes of developmental anomalies, birth defects and genetic diseases.
Other sequencing initiatives are less focused on obtaining highly accurate and/or complete genomes for comparative genomic work. Many are essentially “fishing expeditions,” looking to see if something interesting shows up, or to identify molecular markers that can subsequently be used for management and conservation efforts. For example, the recently published white shark genome found that olfactory genes were not as abundant as expected given white sharks’ good sense of smell, and that white sharks have a higher proportion of transposable elements – DNA sequences that can move from one location on the genome to another – than is typical.
Such projects are usually much less expensive, since they are not designed to obtain high-resolution genomic maps with complete coverage of the genome. Unfortunately, they have limited utility for downstream research. They are generally too incomplete to be useful for developmental biologists, and are of limited use for understanding the genotype-phenotype map.
Nonetheless, they do serve to spur public interest in the burgeoning field of genomics, which is already having a big impact in fields ranging from basic biology to applied personalized medicine. As more high-resolution genomes are gathered and compared, we can expect that our understanding of the architectures underpinning different life forms will expand exponentially.