

La surveillance des mutations du coronavirus susceptibles d’aggraver la pandémie ou d’atténuer l’efficacité vaccinale est un enjeu majeur pour le pilotage des mesures de contrôle sanitaire.
Augmenter les capacités de séquençage des virus, qu’ils soient prélevés chez des patients ou dans des échantillons environnementaux ou animaux, puis partager ces données est donc indispensable pour comprendre les origines des épidémies actuelles et pour mieux se préparer à celles qui émergeront demain. Mais pas seulement.
Accélérer la recherche, le diagnostic et la mise au point de traitements
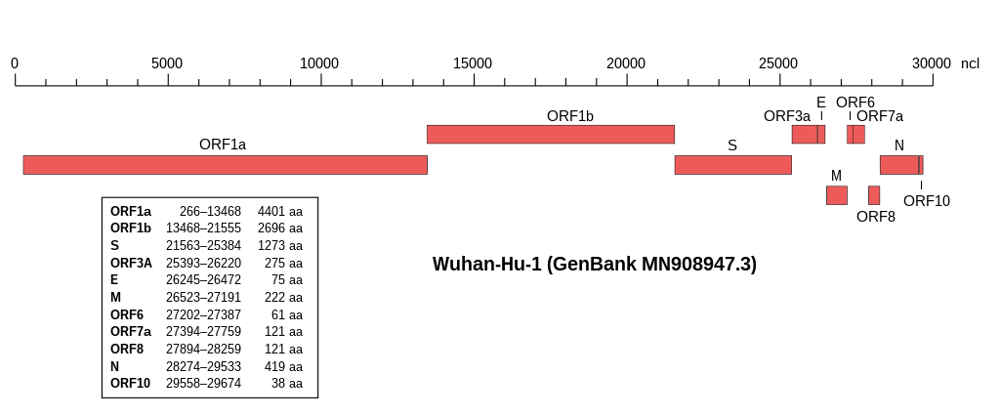
Avec les techniques d’aujourd’hui, il est possible à partir d’un prélèvement respiratoire ou nasopharyngé d’obtenir la séquence (le « texte génétique ») d’un virus responsable d’une nouvelle maladie en un à cinq jours – on parle de séquençage. La séquence des 30 000 lettres qui forme le génome du SARS-CoV-2, agent du Covid-19, a été déterminée fin 2019 et rendue publique le 11 janvier 2020.
Son analyse a permis de comprendre que l’épidémie en cours dans la ville de Wuhan était liée à un coronavirus inconnu mais apparenté au SARS de 2003. Forts de ces informations, les virologistes en ont déduit qu’il devait probablement se transmettre par gouttelettes, aérosols et manuportage et pénétrer dans nos cellules via le récepteur ACE2 présent à leur surface.
La séquence à également permis la mise en place de tests diagnostiques PCR avec une célérité inédite. Ainsi, dès janvier 2020 en Chine, les patients ont bénéficié de tests PCR. De plus, les méthodes de biologie synthétique ont permis aux laboratoires de recherche de produire, sans avoir besoin d’isoler le virus infectieux, certaines protéines du pathogènes pour étudier leur structure et la capacité de neutralisation par des anticorps issus du sang de patients infectés ou vaccinés).
Les virologistes ont quant à eux utilisé la séquence pour synthétiser des clones de ce virus en quelques semaines, afin de pouvoir l’étudier au laboratoire, en infectant des cellules humaines ou des animaux modèles (souris, hamsters, etc.). Leur but était de comprendre comment il perturbe l’organisme, et de tester l’effet de vaccins ou de médicaments potentiels.
Dans une chronologie inédite, le tableau clinique de la maladie Covid-19 avec ses différents symptômes a été dressé par les médecins chinois plusieurs semaines après le séquençage du virus responsable… Et le virus responsable (SARS-CoV-2) de la pandémie a même été nommé avant la maladie (Covid-19) !
Les compagnies pharmaceutiques ont développé des vaccins en un temps record (quelques mois) en utilisant seulement une petite partie du génome : celle qui code la protéine Spike, qui recouvre la surface du coronavirus. C’est elle que notre corps rencontre quand il est infecté par le SARS-CoV-2, et contre laquelle notre système immunitaire produit la majorité des anticorps de défense. La plupart des vaccins actuels visent ainsi à induire l’immunité contre cette protéine.
En conclusion, l’obtention rapide des premières séquences de ce nouveau virus a permis la mise au point rapide de tests de diagnostic, a accéléré la recherche et a facilité le développement d’antiviraux et de vaccins.
Identifier les nouveaux variants problématiques
C’est la première fois dans l’histoire de l’humanité qu’un virus est séquencé autant de fois.
La base de données GISAID (Global Initiative on Sharing Avian Influenza Data), reconnue par la communauté internationale et d’accès libre, comprend actuellement plus de sept millions de séquences de SARS-CoV-2. Comme tous les virus, le coronavirus responsable du Covid-19 mute avec le temps : il accumule environ deux mutations par mois avec parfois des accélérations, comme avec l’apparition des variants Delta ou Omicron.
La plupart des mutations ont peu ou pas d’incidence sur les propriétés du coronavirus. Certaines, toutefois, peuvent avoir un effet sur sa transmission, sa pathogénicité, l’efficacité des vaccins ou des médicaments voire sur l’efficacité des tests de diagnostic. Or, quand on découvre une nouvelle mutation, on ne peut pas déduire directement son effet : il faut analyser le comportement du nouveau variant – à l’échelle de la population humaine (est-ce qu’il rend les personnes plus malades ? Se propage plus vite ?) ou bien au laboratoire sur des animaux ou des cellules en culture.
Les nouveaux outils d’analyse des données de séquence permettent d’identifier l’origine géographique des variants qui ont une diffusion préoccupante autour de foyers épidémiques. Mais également de les classer (Variant of interest ou VOI, et variant of concern ou VOC, selon la terminologie de l’OMS) en fonction de leur cinétique de diffusion et de leur impact potentiel sur la suite de l’épidémie.
Ces analyses indiquent que les mutations ne sont pas réparties aléatoirement le long du génome du coronavirus : elles sont surtout concentrées dans la région codant la protéine Spike.
Certaines de ces mutations permettent au virus d’échapper aux contrôles immunitaires et d’infecter des personnes qui ont déjà attrapé le Covid-19 ou qui sont vaccinées. Cela indique qu’en plus de son rôle capital dans l’entrée du virus dans la cellule, la protéine Spike joue un rôle clef dans l’échappement du virus aux contrôles immunitaires. Les immunologistes ont confirmé ce fait en démontrant qu’elle contient les principaux domaines reconnus par les anticorps capables de neutraliser le virus.

De plus, des études de biologie structurale permettent de visualiser la structure tridimensionnelle de la protéine Spike liée avec les anticorps neutralisants, et d’analyser leurs domaines d’interactions. Or c’est précisément dans ces domaines que le taux de mutation sélectionné est le plus important, ce qui suggère que l’échappement au système immunitaire est un puissant moteur de l’évolution de ce coronavirus. Des modèles informatiques de ces structures 3D permettent de « prédire » l’effet des mutations sur l’échappement immunitaire.
L’étape suivante consiste à prédire comment les différentes mutations observées sur les varient affectent la transmission du virus, la gravité de la maladie et à prédire le plus rapidement possible le comportement d’un nouveau variant.
Dans un contexte où le coronavirus évolue rapidement et échappe à la protection conférée par les vaccins, il devient nécessaire d’adapter ces derniers aux nouvelles souches virales circulantes. Cette stratégie a démontré son efficacité dans le cas de la grippe, dont le vaccin est remis à jour tous les ans en fonction des séquences collectées des virus en circulation. Des vaccins adaptés au variant Omicron sont en cours d’essais cliniques, et nous espérons qu’ils démontreront la pertinence de cette stratégie dans la lutte contre cette infection virale.
Afin que les vaccins soient adaptés aux nouvelles souches à fort potentiel d’échappement immun, le suivi de l’épidémie par séquençage des virus circulants est absolument nécessaire.
En résumé, le séquençage des souches virales en circulation permet d’identifier les variants préoccupants, qui se transmettent plus vite ou échappent au système immunitaire. Il est également une source d’information essentielle pour adapter les vaccins.
Suivre l’évolution de l’épidémie en temps réel
En comparant les séquences, on peut établir des arbres de parenté qui retracent l’ordre d’apparition des mutations dans différentes branches – ces arbres généalogiques sont dits « phylogénétiques ».
Au tout début de la pandémie, le séquençage des virus SARS-CoV-2 montrait toujours des génomes identiques, à deux à cinq lettres près. Ce qui indiquait que l’ancêtre commun à tous ces virus remontait à quelques mois. Aujourd’hui, le variant Omicron comporte plus de 60 mutations par rapport à la forme initiale observée à Wuhan en décembre 2019.
L’analyse des séquences issues des premiers patients Covid-19 détectés aux États-Unis début 2020 a montré qu’elles ressemblaient plus aux séquences obtenues en Europe (qui avaient commencé à diverger de la séquence de Wuhan) qu’à celles observées en Chine. Ces analyses ont permis de conclure qu’il y avait eu plusieurs introductions indépendantes du virus dans le pays, à partir de voyageurs venus d’Europe, et que la fermeture des frontières avec la Chine avait limité les introductions depuis ce pays.

Le site nextstrain analyse en temps réel toutes les nouvelles séquences obtenues par les pays qui partagent leurs données et permet de visualiser leur évolution. Ce partage mondial est extrêmement important, car c’est en recoupant toutes les données disponibles qu’on peut extraire le plus d’informations possible – et notamment identifier les variants préoccupants.
L’analyse des séquences à plus petite échelle permet également de reconstituer les chaînes de transmission locales. Par exemple, en janvier 2022 à Hongkong, une vendeuse et une cliente d’un magasin animalier ont été infectées par le variant Delta, alors que celui-ci ne circulait pas dans la région. La comparaison des séquences virales des deux patientes a révélé cinq différences, excluant une transmission directe de l’une à l’autre. Le séquençage de prélèvements effectués sur les animaux vendus dans le magasin a montré que le coronavirus provenait en fait de hamsters syriens importés des Pays-Bas, et que vendeuse et cliente avaient été infectées indépendamment par deux hamsters porteurs de souches virales légèrement différentes !
Enfin, il faut noter que le virus est maintenant détectable à partir des eaux usées. Par ce moyen, il est possible de repérer une flambée épidémique quelques semaines avant d’observer la montée des cas à l’hôpital. Ces techniques sont prometteuses, même si le séquençage des virus à partir de ce type d’échantillon reste complexe car le signal est faible et donc sujet à beaucoup de bruit de fond.
Retracer les origines des épidémies
L’analyse des séquences est aussi un outil précieux pour rechercher les origines des épidémies.
Même si au moment de l’émergence d’une nouvelle maladie, on ne dispose pas des virus ancêtres les plus proches, un travail d’échantillonnage et de séquençage minutieux permet généralement de préciser l’histoire évolutive d’un virus. En 2020 et 2021, les autorités chinoises ont analysé plus de 80 000 prélèvements d’animaux sauvages, d’animaux d’élevage et d’échantillons environnementaux et n’ont malheureusement pas réussi à trouver un virus progéniteur de l’épidémie de Covid-19.
Par contre, le séquençage d’échantillons anciens prélevés sur des chauves-souris dans le sud de la Chine ou dans les pays limitrophes comme le Laos, a mis en évidence des cousins du SARS-CoV-2 avec une similarité de séquence allant jusqu’à 97 %. En se basant sur leur rythme de mutations, on a pu en déduire qu’il y a environ 20 à 40 ans, l’ancêtre du SARS-CoV-2 circulait dans les populations naturelles de chauves-souris.
Toutefois, les estimations temporelles sont imprécises. D’une part parce que le rythme de l’évolution n’est pas linéaire, d’autre part parce que ces virus sont sujets à de multiples recombinaisons : lorsque plusieurs coronavirus différents co-infectent un même animal, ils ont tendance à échanger des fragments de leur génome. Sporadiques, ces événements jouent un rôle clé dans leur évolution et dans le franchissement de la barrière d’espèce.
Malheureusement, cela complique les analyses phylogénétiques. D’où l’incertitude importante dans l’estimation du temps séparant deux virus proches. Ce qu’il s’est passé en 20 à 40 ans entre le virus circulant chez les chauves-souris et son avatar détecté chez les humains fin 2019 reste un mystère.
La résolution de l’énigme de l’origine du SARS-CoV-2 nécessiterait un échantillonnage plus important des virus circulants actuellement dans la faune sauvage, dans les élevages et dans les laboratoires. Contrairement à d’autres virus comme le HIV qui persistent pendant des années dans les individus infectés, ceux responsables d’infection aiguë comme le SARS-CoV-2 ne restent que quelques jours dans leurs hôtes, ce qui complique ces opérations.
De plus, comme ces virus sont capables d’infecter plusieurs espèces de mammifères, l’infection des animaux par les humains (on parle alors de zoonose inverse) complique les analyses. L’idéal serait de disposer de deux types d’échantillons anciens : prélevés sur des patients (sang, urine, prélèvements respiratoires, etc.) puis congelés pour rechercher les premiers infectés (patient Zéro), et sur des animaux congelés pour détecter les virus ancêtres présents avant l’émergence du SARS-CoV-2.

Si les échantillonnages sont suffisants et l’accessibilité aux données garantie, alors les analyses des séquences, couplées aux informations spatio-temporelles, permettent de mieux localiser les origines géographiques des épidémies et de mieux comprendre comment le virus a franchi la barrière d’espèce et commencé à infecter notre espèce.
Ces conditions ne sont malheureusement pas réunies dans le cas de l’émergence du SARS-CoV-2. L’enjeu pour le futur est de mettre en place des collectes et stockages systématiques de prélèvements effectués chez des patients, et d’échantillons environnementaux et animaux. Ceci pour une meilleure surveillance épidémiologique et une compréhension des mécanismes d’émergence virale.
Enjeux pour le contrôle des futures épidémies
Avec la baisse des coûts et une sensibilité accrue, le séquençage est devenu en quelques années une méthode puissante pour le suivi des épidémies et la prédiction de leur trajectoire. L’épidémie de Covid-19 illustre l’intérêt du séquençage accru des virus circulants pour une meilleure gestion des épidémies comme la surveillance de nouvelles émergences.
Mais notre difficulté à identifier son origine indique que des efforts supplémentaires sont nécessaires. Plusieurs axes pourraient être renforcés :
Une collecte et un séquençage plus systématique d’échantillons environnementaux (eaux usées…) et d’origine animale, d’élevage et sauvages, permettraient de mieux suivre l’émergence des pathogènes. En ce qui concerne l’échantillonnage des animaux de la faune sauvage, les risques de contamination des expérimentateurs et les risques liés à la construction de virus chimériques doivent être mieux pris en compte.
Des outils devraient être développés afin d’exploiter les résultats des séquençages effectués dans le monde entier. En effet, ces données contiennent systématiquement des traces de contaminations environnementales (par l’air ambiant du laboratoire par exemple) et, actuellement, une fraction non négligeable des « reads » issus du séquençage d’un échantillon quelconque (plante, bactérie, etc.) correspond au SARS-CoV-2.
Le dépôt des informations de séquençage et le développement d’outils informatiques permettant leur analyse sont nécessaires pour la gestion des futures pandémies. Le libre accès aux données est capital afin d’éviter tout contrôle par des entreprises privées ou des États.
Dans la mesure où l’émergence du SARS-CoV-2 dans les populations humaines pourrait impliquer un accident de recherche, une autorité internationale devrait avoir la copie des séquences de virus manipulés en laboratoire. Les laboratoires eux-mêmes devraient être dotés de boîtes noires biologiques.
Le séquençage est devenu un outil incontournable pour la surveillance épidémiologique et le contrôle des futures épidémies. Il faut se féliciter que l’Organisation mondiale de la santé (OMS) considère son développement comme une priorité internationale. Pour compléter ce système de surveillance, nous pensons que l’OMS devrait se doter d’une task force spécialisée intervenant systématiquement sur les lieux d’épidémies nouvelles et dont le rôle serait de collecter des échantillons pour en comprendre les causes.