



A mediados del siglo XX, los doctores suizos Andrea Prader, Alexis Labhart y Heinrich Willi describieron a nueve pacientes que presentaban un cuadro clínico similar, caracterizado por la obesidad, talla baja, manos y pies pequeños, así como por alteraciones en el aprendizaje. Además, todos ellos sufrían hipotonía o bajo tono muscular desde el nacimiento.
A partir de las descripciones aportadas por estos médicos, se definió esta entidad clínica con la nomenclatura de síndrome de Prader-Willi. Aunque actualmente, el nombre de Labhardt no aparece en el síndrome, sí que lo hizo cuando salieron las primeras monografías publicadas en los años sesenta.
Se estima que hay entre 350 000 y 400 000 personas con este síndrome en el mundo y que tiene una prevalencia al nacimiento de 2,8 por cada 100 000. Por eso se encuentra dentro de las denominadas enfermedades raras o poco frecuentes, ya que presenta una prevalencia inferior a 5 casos por 10 000 habitantes.
Escrito en el ADN
El diagnóstico de este síndrome debe hacerse mediante un estudio genético, pero orientado a descubrir cuál de las principales causas descritas es la que acontece en cada niño.
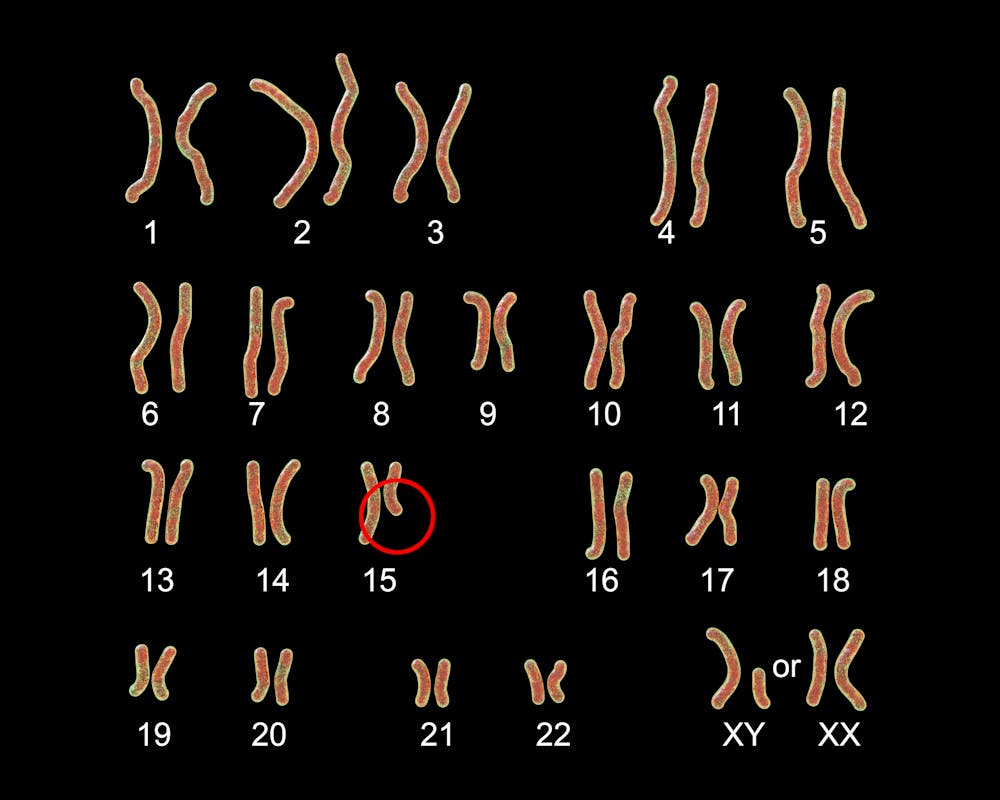
Hoy en día se conoce que este síndrome aparece cuando sucede algo anómalo en un parte del cromosoma 15 que hace que los genes del padre no se expresen adecuadamente. El origen del problema puede ser que le falta un trozo del cromosoma 15 del padre –deleción– o que se han heredado dos copias de la madre –disomía uniparental–.
La causa más frecuente es la deleción de la región 15q11.2-q12, aunque se han descrito casos con una deleción que abarca una zona más extendida, además de otros tipos de alteraciones genéticas como traslocaciones (partes el cromosoma localizadas en otro cromosoma).
El diagnóstico definitivo, por tanto, exige un estudio genético dirigido. Estos cambios también pueden verse con las nuevas técnicas de secuenciación masiva del exoma y del genoma.
Cuando la comida se convierte en obsesión
El síndrome de Prader-Willi no distingue ni sexos ni etnias. A medida que el niño o la niña crece, la sintomatología puede ir evolucionando. La mayor parte de las manifestaciones clínicas no desaparecen, sino que se van sumando nuevos síntomas.
En el periodo fetal y neonatal, podemos encontrar una disminución de los movimientos esperables del feto, succión y llanto débiles. También pueden aparecer problemas de caderas, con posible dislocación de la misma, y criptorquidia, que ocurre cuando uno o ambos testículos no descienden completamente hasta el escroto. La hipotonía o escasa tensión muscular suele mostrar signos de mejora a partir de los 2-3 años.
Hasta los tres años, los afectados pueden presentar una talla baja con respecto a su edad y una cara característica con ojos en forma de almendra, estrabismo, boca triangular, manos pequeñas o baja pigmentación de piel y cabello. Además, empiezan a observarse retrasos madurativos en el crecimiento, a nivel psicomotor y en el lenguaje.
Otro rasgo característico es la obesidad incipiente que, a partir de los tres años y hasta la adolescencia, tiene su origen en una hiperfagia o ingesta de comida en exceso. Llega al extremo de que los afectados buscan la comida de forma obsesiva. Todo apunta a que la raíz del problema se encuentran en que los mecanismos cerebrales que controlan la saciedad están alterados.
A todo ello, hay que añadir posibles alteraciones del comportamiento entre las que se incluyen rabietas con arranques violentos. Por si fuera poco, aparecen problemas de inestabilidad emocional, inflexibilidad, tendencia a la confrontación o comportamientos obsesivo-compulsivos.
En la edad adulta, la hiperfagia continúa y aparecen otras enfermedades relacionadas con la obesidad, osteoporosis, así como trastornos psicóticos y afectivos.
Alteraciones cognitivas
Desde el punto de vista cognitivo, el síndrome causa discapacidad intelectual, que puede reflejarse en dificultades para el logro de la lectoescritura, realizar cálculos matemáticos, así como para el razonamiento abstracto.
La falta de flexibilidad cognitiva y el bajo nivel de inteligencia emocional pueden generar dificultades en el área social y predisponer al desarrollo de rabietas, tan característico en estos cuadros. Resulta clave un abordaje multidisciplinar, desde la perspectiva de la atención temprana, para trabajar los aspectos cognitivos y desarrollar la inteligencia emocional.
En esta misma línea, conviene ser constantes en el apoyo a la salud mental de estos pacientes, incluyendo criterios estrictos en el tema de las comidas, actitudes saludables y soporte conductual.
Impacto en la familia
La comunicación de un diagnóstico genético de estas características genera un impacto psicológico significativo. No solo en el paciente, sino en la familia.
Los padres y madres suelen sentir que se les viene el mundo abajo. Es común que aparezcan sentimientos de miedo, angustia e incertidumbre. En el caso del síndrome de Prader-Willi, la sospecha diagnóstica suele darse durante los primeros meses de vida, con especial impacto en el neurodesarrollo y la alimentación.
A medida que van creciendo, las dificultades cognitivas, la presencia de rabietas y las conductas vinculadas a la búsqueda obsesiva de comida son aspectos que suelen estresar mucho a los cuidadores. Por ello, dentro del abordaje interdisciplinar debe tenerse en cuenta la importancia de cuidar al cuidador.
Para las familias es clave obtener apoyo de otras familias, de asociaciones o incluso de un profesional experto, para sentirse entendidas y escuchadas. Más aún en las primeras etapas de la enfermedad, cuando se acaba de recibir el diagnóstico y surgen emociones que requieren un mayor acompañamiento y contención.
El abordaje multidisciplinar es la clave
Estamos ante un síndrome que afecta principalmente a la regulación hormonal de funciones tan importantes como el apetito, el desarrollo sexual y glándulas como el tiroides.
Muchos de los problemas que padecen estos pacientes se deben a su continuo deseo de comer, lo que les lleva a la obesidad y a complicaciones secundarias como la diabetes. En algunos casos, se aplican tratamientos con hormonas sexuales y del crecimiento, para lograr un desarrollo más acorde a lo esperado. La posible presencia de criptorquidia (no descenso de los testículos) puede requerir tratamiento quirúrgico en algunos casos.
Otros trastornos, como los problemas relacionados con el sueño, tanto somnolencia como apnea del sueño, precisan una pronta identificación y el soporte adecuado dependiendo de la gravedad. Los trastornos óseos en la columna y de la cadera, debidos a osteoporosis, pueden requerir vigilancia y tratamiento suplementario con calcio. Y los trastornos visuales, atención de especialistas.
Pero, sin duda, los tratamientos más importantes serán los dirigidos a la vigilancia y control de la conducta alimentaria compulsiva.
Al tratarse de un problema genético complejo, de momento no es posible contar con un tratamiento único y curativo para esta enfermedad. Sin embargo, durante los últimos años se han registrado 109 ensayos clínicos, 55 de los cuales ya se han completado y se está a la espera de resultados. Algunos de estos estudios se centran en fármacos que actúan sobre el apetito o los efectos secundarios de esta enfermedad en el desarrollo de los niños.
Es fundamental seguir investigando y manteniendo la esperanza en el logro de un tratamiento que ayude a curar o paliar la sintomatología. Mientras llega ese momento, es necesario que las familias tengan recursos de apoyo que les ayuden a mejorar su calidad de vida y a crecer integrados en la sociedad.